A DNS kettős hélix szerkezetének megismerése, és ezen keresztül a genetikai információ megfejtése méltán tartható a 20. század második fele legnagyobb felfedezésének. A felismerés gyakorlati haszna nemcsak a biotechnológia eredményein át vált alig felbecsülhető értékké az ipar és mezőgazdaság számára, hanem a humán gyógyászatban is hatalmas lehetőségeket nyitott meg. Ezen belül kiemelkedően fontosnak tartható azoknak a gyógyszereknek a megismerése, melyek hatásukat a nukleinsavakkal való kölcsönhatás révén fejtik ki. A genomika azon ágazataival ellentétben, ahol a cél a hasznos genetikai információ megőrzése illetve átvitele, a nukleinsav támadáspontú gyógyszerek adott - emberi szervezetre káros - információk kifejeződését akadályozzák meg.
A humán illetve hasznos élőlényekre káros gén örökletes illetve szerzett úton juthat a szervezetbe. Utóbbiak között a kórokozók által bevitt, a hibás endogén anyagcsere folyamatok útján képződő, valamint a környezeti ártalmak révén keletkező változások a legjelentősebbek. A káros gének kifejeződése számos patológiai eseményhez vezet, melyek közül a rákos megbetegedések és a vírus- valamint a baktériumfertőzés okozta ártalmak genetikai alapjai a legismertebbek. Bár a gyógyszeres terápia nyilvánvalóan szoros kapcsolatban van az említett területekkel, jelen igen vázlatos összeállítás a megbetegedések kezelésében alkalmazott nukleinsav támadáspontú anyagok molekulár-farmakológiai alapjaival foglalkozik. Az anyagcsoport fontosságát mutatja, hogy a Magyarországon forgalomban lévő összes gyógyszert ismertető Gyógyszerkompendium 2002. évi kiadásában szereplő daganatellenes szerek több mint 60%-a, az antivirális anyagok több mint 70%-a közvetve vagy közvetlenül nukleinsav támadáspontú vegyület. A világ gyógyszerforgalmában is hasonló arányok mutathatók ki.
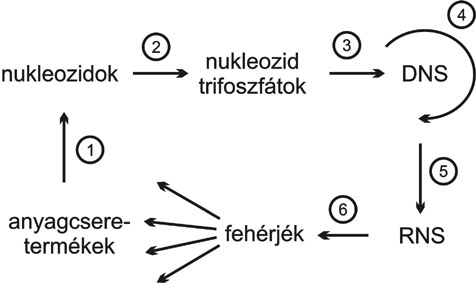
A nukleinsav információk sejteken belüli kifejeződésének megakadályozására számos, az 1. ábrán feltüntetett biokémiai folyamat inhibiálása útján van lehetőség.
1. ábra DNS bioszintézisének és információ kifejeződésének vázlata
A DNS bioszintézisét gátolhatjuk olyan módon, hogy megakadályozzuk nukleozid építő egységek keletkezését (1. ábra (, (, ( folyamat). Ezzel ab ovo lehetetlenné válik a káros gént tartalmazó DNS szintézise. Ezen a hatásmechanizmuson alapuló anyagok (például: Methotrexat, Mercaptopurin) a daganatellenes vegyületek igen jelentős, a gyógyszeres terápiában széles körben alkalmazott csoportját alkotják. Mivel ezek csak következményeiben érintik a nukleinsav funkciók érvényesülését, az átalakulásokat gátló inhibitorok nem képezik jelen összeállítás tematikáját. A tárgyalt vegyületek közé azok tartoznak, melyek a nukleinsavakkal végbemenő közvetlen kölcsönhatások révén vagy azokba beépülve inhibiálják a genetikai információ átadási (replikáció, transzkripció, transzláció; 1. ábra (, (, () folyamatokat. A kölcsönhatások természete alapján a gyógyszereket az alábbi fő csoportokba soroljuk
* DNS-el kovalens kötést létrehozó vegyületek
* DNS-el interkalációs komplexet képző szerek
* DNS-be beépülő nukleozid analógok
* Nukleinsavakkal hibridet alkotó oligonukleotidok (antiszensz oligomerek)
Az első két csoport csaknem kizárólag a rákos betegek kezelésében, a harmadik nagyobb részben a vírus, kisebb, de számottevő mértékben a daganat-terápiában, míg a negyedik mindkét hatástani területen alkalmazott gyógyszercsalád. A baktérium kemoterpiában használatos vegyületek speciális komplexképzők. A folyamatokban a DNS-en és a gyógyszeren kívül enzimek is részt vesznek.
A felsorolt vegyületcsoportok mindegyikében folytak illetve folynak kutatások különböző magyarországi laboratóriumokban.
A szelektivitás problematikája
A nukleinsav támadáspontú vegyületek elsődleges gyógyszeralkalmazási feltétele a szelektivitás. Ez a kritérium, mely valamennyi gyógyszerhatástani területen alapfeltétel, a tárgyalt vegyületek körében különleges fontossággal bír. A káros és hasznos gének szerkezeti tulajdonságai ugyanis igen hasonlóak. Alapvető szerkezetbeli különbségek a nukleinsavak funkciójából adódó szekvenciális különbözőségben rejlenek. Ezen az alapon a fenti csoportosítású szerek két részre oszthatók. Az egyikbe a szekvenciára kevésbé érzékeny, a másodikba a nagy szekvencia-specificitású vegyületek tartoznak. Az első két vegyületcsoport szekvencia-specificitása jelenlegi ismereteink alapján mérsékelt illetve kicsiny. Ezen első generciós nukleinsav támadáspontú hatóanyagok gyógyszerként való alkalmazhatóságát elsősorban egyéb szelektivitási tényezők adják, melyek közül a legfontosabbak a bioaktivációs és bioinaktivációs folyamatok. Ezek tudatos alkalmazásával létrehozott kemoterápiai szerek a második generációs nukleinsav célpontú hatóanyagok csoportját képezik.
A szelektivitást növelő biokémiai átalakulásokat specifikus enzimek katalizálják. Jelentőségben kiemelkednek közülük azok, melyek közreműködésével válik lehetővé a DNS többszörösen felcsavarodott szerkezetéből a replikációra és transzkripcióra alkalmas forma kialakulása (topoizomerázok, giráz). Ezen folyamat inhibitorai között számos fontos daganat és baktérium ellenes gyógyszer található.
A harmadik csoportba sorolt vegyületek szelektivitása lényegesen nagyobb. A nukleinsavakba történő beépülés ebben az esetben is enzimek közreműködésével jön létre. A szelektivitás mértéke az adott enzim-tumor illetve vírusfertőzött sejt specificitásától függ.
Fenti felsorolásban szereplő negyedik vegyületcsoportot oligonukleotidok képezik, melyek a Watson-Crick-féle bázis összekapcsolódási szabályoknak megfelelően képesek adott nukleinsav szakasz kimagasló specificitású felismerésére és hibridizáció révén a káros információ-továbbítás inhibiálására. Ezen elv felhasználásával két évtizede kezdődött gyógyszerkutatás a legjelentősebb irányzatának tekinthető a nukleinsav támadáspontú anyagok körében, azok harmadik generációs típusát alkotva. A vegyületek alkalmazási területe jelenleg elsősorban a kemoterápia, de perspektivikusan egyéb hatástani területeken is vezető, legalábbis kimagasló szerephez juthatnak. Ennek megfelelően jelen - a terjedelem megszabta lehetőségek miatt igen vázlatos - összehasonlításban is kiemelten foglalkozunk az antiszensz oligonukleotidokkal.
DNS-el kovalens kötést képző vegyületek
A daganatgátló hatású gyógyszerek legrégebbi, a mai napig egyik legjelentősebb csoportját azok a vegyületek alkotják, melyek a DNS-t felépítő heterociklusos bázisokkal kovalens kötést hoznak létre. Már a múlt század első harmadában megfigyelték, hogy az alkilező szerek kromoszóma-károsodást okoznak. A harmincas évek végén kimutatták, hogy a harci gáznak kifejlesztett b,b-diklórdietilszulfid (mustárgáz) (2. ábra 1.) sejtproliferáció gátló hatású. Rákellenes gyógyszerként a vegyület nem jöhetett számításba a daganat- és egészséges sejtek közötti igen kis szelektivitása miatt. Hamarosan kiderült, hogy a kénatom NH-ra történő cseréje a szelektivitást igen jelentős mértékben megnöveli. A b,b-diklórdietilamin (2.) azzal az előnnyel is rendelkezik, hogy az NH-csoport hidrogén atomja legkülönbözőbb csoportokkal helyettesíthető, így tetszőleges számú származék készíthető el. Elsőként az N-metil-b,b-diklórdietilamint (3.) vezették be a gyógyászatba. 1946-ban amerikai kutatók (Jacobson et al., 1946) már harminchárom hónapos daganatterápiai tapasztalatról számoltak be, több szarkoma esetén igen jó eredménnyel. Felhívták a figyelmet, hogy a különböző tumorok igen különböző érzékenységűek, és az egészséges sejtek károsodása nagyfokú, ezért igen kis (0,1 mg/kg) dózist használtak. A mustárnitrogének alkalmazása áttörést jelentett a rákgyógyászatban. Bebizonyította, hogy a daganatos betegségek gyógyszeresen is kezelhetőek.
Angol kutatók (Haddow, Ross, Lawley, Brookes) a nitrogenmustard családot tovább fejlesztették. Elsősorban olyan származékokat szintetizáltak és vizsgáltak, melyekben a nitrogenmustard metil csoportja helyett fenil illetve szubsztituált fenil csoportot építettek be a molekulába, azaz az N,N-bisklóretil-anilin származékait szintetizálták. Legsikeresebb citosztatikus hatású vegyületnek az bizonyult, melyben a b,b-diklórdietilamino csoporthoz kapcsolt fenil gyűrű p-helyzetben g-karboxi-propil csoportot tartalmaz (4.). A vegyületet mai napig használják rákellenes gyógyszerként klórambucil néven.
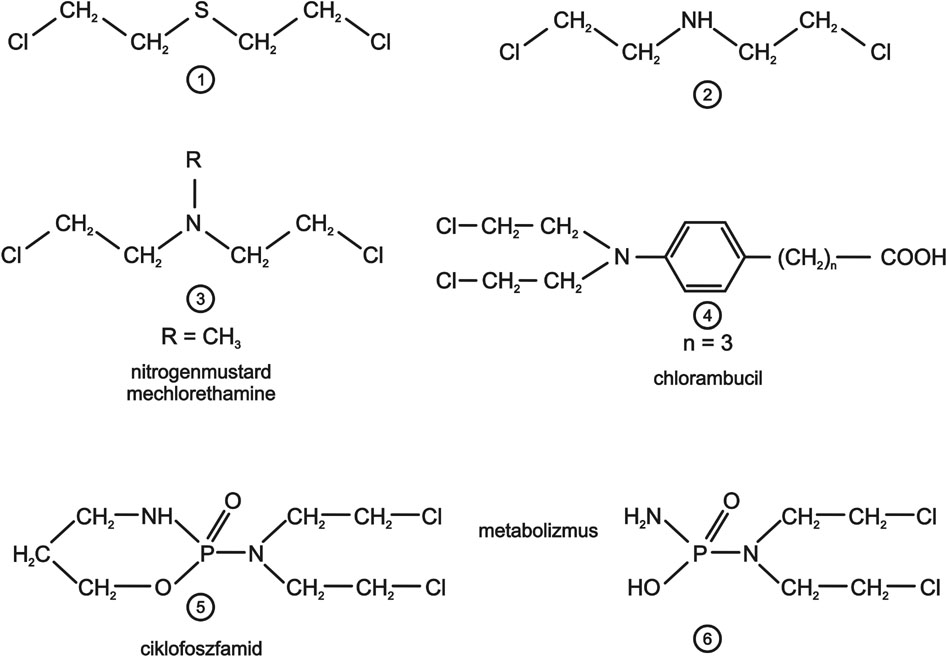
2. ábra. Bifunkciós biológiai alkilező szerek I. Kénmustár és nitrogénmustár származékok
Alexander Haddow az experimentális daganatkemoterápia egyik megalapítója, a vegyületek hatásmechanizmusáról egy 1949-ben tartott konferencia írásos összefoglalójában azt a véleményt fejtette ki, hogy az ilyen típusú szerek kromoszómakárosító hatásáért a bennük lévő proteinek alkilezése, mégpedig keresztkötést létrehozó kémiai reakciója felelős.
A hatásmechanizmus megismerésében alapvető változást jelentett a DNS kettős hélix megismerése. Szinte kézenfekvő feltételezésként merült fel, hogy a keresztkötés a kromoszómában lévő DNS két szála között alakuljon ki. Erre vonatkozó bizonyítékot az angol iskola munkatársai Peter Brookes és Philip Lawley a kettős spirál felismerése után néhány évvel (Brookes - Lawley, 1961) publikálták. A kettősen alkilezett DNS lebontási reakcióival igazolták, hogy keresztkötés nagymértékben a szemközti szálakon helyet foglaló guanin bázisok N7 nitrogén atomjai között jön létre. (3. ábra) A felismerés három szempontból is kiemelt jelentőségű. (a) Bizonyította DNS meghatározó szerepét a gyógyszerek hatásában; (b) A DNS szerkezeti tulajdonságainak ismeretében új szerek tervezésének lehetőségét nyitotta meg; (c) Kimutatta a szelektivitás korlátozott lehetőségeit az alkalmazott vegyületek körében.
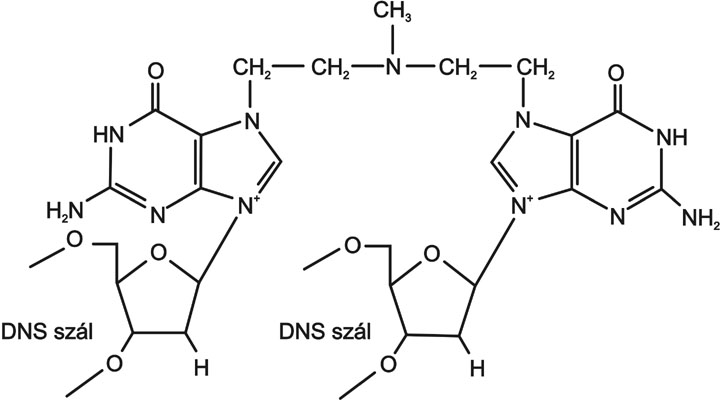
3. ábra Mustárnitrogénnel keresztkötött DNS
A bifunkciós biológiai alkilező szerek DNS keresztkötési reakciói a mai napig kutatási témát jelentenek, amit indokol, hogy a vegyületek, beleértve a következőkben tárgyalt származékokat, jelenleg is a daganatellenes vegyületek nagy százalékát alkotják. A vizsgálatok fő iránya a DNS szerkezeti tulajdonságainak bővülő ismerete alapján annak a kérdésnek a tisztázása, milyen összefüggés van nukleinsav szerkezeti paraméterek és az alkilező vegyületek szerkezeti adottságai között (a). Ezek a tulajdonságok, milyen korrelációban vannak a tumorellenes szelektivitással (b). A több évtizedes, sok laboratóriumban végzett vizsgálatok az alábbi általános összefüggések megállapítását tették lehetővé.
* A keresztkötés nagymértékben a DNS nagy árkában helyet foglaló bázisspirálok között jön létre.
* A két szál N7-guanin atomjainak összekötése G-C irányú.
* Nagyobb valószínűséggel jön létre keresztkötés a halmozott G tartalmú szekvenciák esetén.
* Az alkilező centrumokat összekötő hosszabb egység esetén nem a szomszédos, hanem az 1,3-helyzetű guanin bázisok között létesül keresztkötés.
Említett DNS szekvenciális szerkezeti tulajdonságok bizonyos gének érintettségét kizárják ugyan, de a tumor-szelektivitás alapvető kérdéseire nem adnak választ. Az egyéb szelektivitást befolyásoló tényezőket - abból a szempontból, hogy bennük a DNS milyen szerepet játszik - két részre oszthatjuk:
* A target DNS-t érintő átalakulások
* A hatóanyag DNS-től független tumor illetve kórokozó károsított sejtspecifikus reakcióik.
A biológiai alkilező vegyületek és DNS közötti reakciók szelektivitására legrégebbi magyarázat - jobb híján - annak a feltételezése, hogy a gyorsabban osztódó sejtekben nagyobb az aktivált állapotú DNS koncentrációja. Mivel tisztán kémiai folyamatról van szó, a reakciókinetikai paraméterek különbözősége specificitással jár együtt.
Előbbinél lényegesen fontosabb, kísérletileg is bizonyított szelektivitási tényező a DNS repair-beli (károsodott DNS-t javító mechanizmus) különbség a daganat és egészséges sejtek között. A tumor sejtekben ez a folyamat lényegesen kisebb mértékben megy végbe, legalábbis a kezdeti szakaszban. Idővel a daganatos sejtek is "megtanulják" a repairt, ami az alkilező szerekkel szemben kialakuló rezisztencia egyik fő okozója. A rezisztencia problematikája egyébként nemcsak ennek a vegyületcsoportnak, hanem egész kemoterápiai területnek központi kérdése. Mindenképpen részletesebb tárgyalást érdemelne meg, de jelen összeállítás a terjedelem korlátai miatt ezt nem teszi lehetővé.
Fentieken túlmenően a DNS alkilezhetősége függ a benne levő nukleofilek (elsősorban guanin N7 atom) pH-függő reaktivitásától, különböző endogén anyagokkal való kölcsönhatástól, a DNS hélixstruktúrán túlmenő speciális rendezettségtől, melyekben a tumoros és normál sejtek között - ma még sok vonatkozásban ismeretlen - különbségek vannak.
A DNS-től független átalakulások szelektivitására már utaltunk. Az említett bioaktivációs és bioinaktivációs folyamatokon kívül jelentős, esetenként meghatározó faktorok: a transzport folyamatok specificitása, továbbá az előbbi folyamatokban nem érintett fehérjék és egyéb anyagcseretermékkel létrejött kölcsönhatások szelektivitása. Hangsúlyozandó, hogy a gyógyszerhatás oki tényezője ezekben az esetekben is a káros DNS információ kifejeződésének megakadályozása. Az említett folyamatok a tumor (illetve vírus) specificitásban nyernek fontos, sok esetben döntő szerepet.
A bioaktiváció az alapja az egyik leggyakrabban alkalmazott, széles hatásspektrumú, a magyar klinikai gyakorlatban is kiemelt jelentőségű gyógyszer, a ciklofoszfamid (5.) hatásának is. A vegyület bár tartalmazza a b,b-diklórdietil-amino molekularészt, gyakorlatilag hatástalan. Használatát az tette lehetővé, hogy kétfajta enzim hatására foszforamid-mustárrá (6.) alakul (2. ábra). A metabolitikus átalakulás tumorspecificitása szelektív hatásban jelentkezik, amit tovább erősít az a tény, hogy gyógyszer az egészséges sejtben egy harmadik enzim katalizálta folyamatban specifikusan bioinaktiválódik (Jeney, 2001).
A ciklofoszfamid tipikus prodrog, melyre a vegyület hatásmechanizmusának felderítése útján derült fény (empirikus prodrog). A prodrog-elv tudatos gyógyszerkémiai alkalmazását a továbbiakban több vegyületcsoportnál tárgyaljuk. Ennek legfejlettebb formái (rövid összefoglalás: Kopper, 2002), mikor a bioaktivációt katalizáló enzimet szelektíven juttatják a tumorsejtbe egy alkalmas vektorral (GDEPT - gene directed enzyme prodrug therapy) vagy tumorsejtspecifikus antitesthez kötik (ADEPT - antibody directed enzyme prodrug therapy).
A mustárnitrogének és DNS közötti reakciók megismert törvényszerűségeire alapozva egyéb bifunkciós alkilező szerek gyógyszerré fejlesztésére került sor. Ilyenek a dimeziloxi alkánok (7.), diepoxidok (8.,13.), nitrozokarbamidok (9.), bróm vegyületek (11.,12.).
A mustárnitrogén származékok körében Szekerke Mária a nitrozokarbamidok területén Medzhradszky Kálmán és Vargha Helga (1987) szintetizált elvi meggondolások alapján peptidekhez kötött citosztatikum prodrogokat. Munkásságuk világviszonylatban is úttörőnek tekinthető.
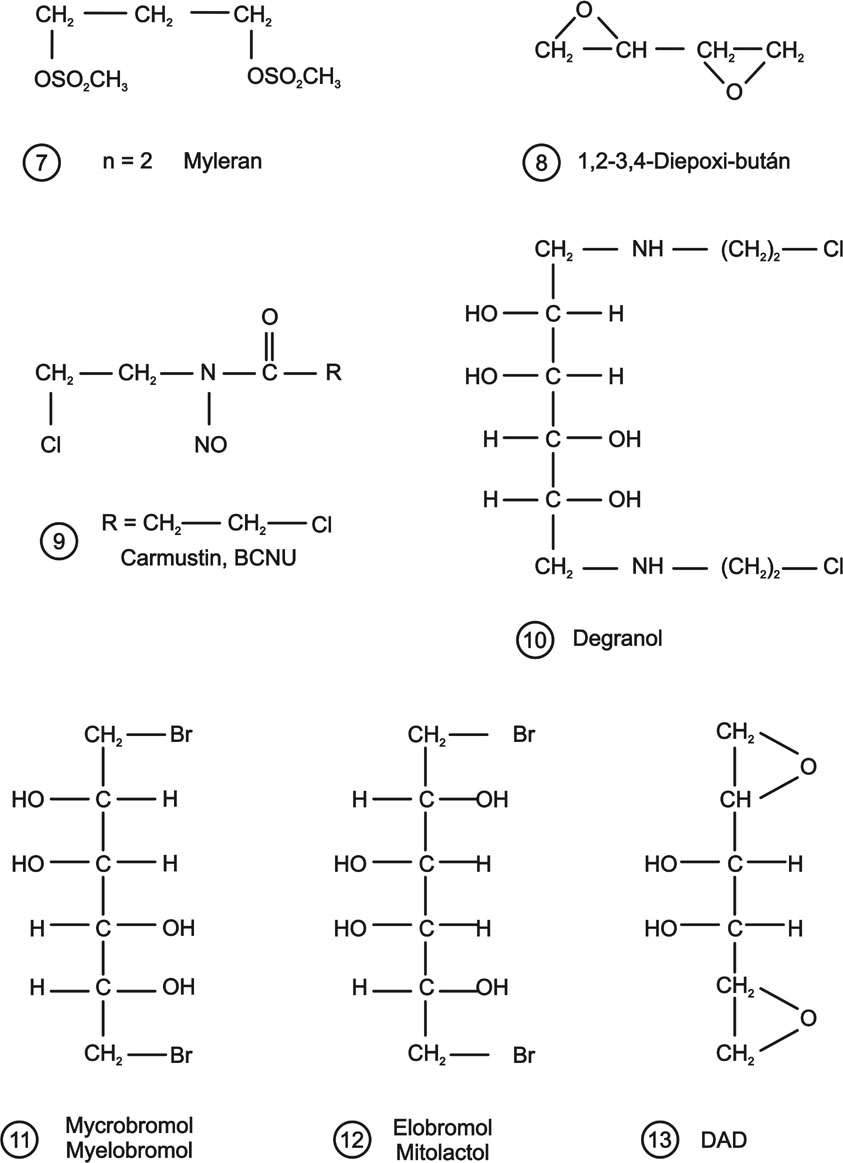
4. ábra Bifunkciós biológiai alkilező szerek II. Mezilátok, epoxidok, cukoralkohol származékok
Mind potenciális rákgyógyászati értékük, mind a hazai kutatási eredmények miatt külön csoportként tárgyaljuk a cukoralkohol származékokat. Első képviselőjük a Varga László és munkatársai által kifejlesztett Degranol (10.) 1,6-helyzetben alkilező b-klóretil-amino csoportot tartalmaz.
Farmakológiailag, klinikailag és molekuláris hatásmechanizmusát tekintve legszélesebb körben tanulmányozott vegyületek az 1,6-dibróm-hexitek (11., 12.). A mannit származék Myelobromol, az 1,6-dibróm-dulcit Elobromol néven vált ismert gyógyszerré. Kidolgozói a Chinoin, az Onkológiai és az Onkopatológiai Intézet Institóris László és Eckhardt Sándor vezette kutatócsoportjai Németh László és Somfai Zsuzsa igen gyümölcsöző közreműködésével. A vegyületek szerkezettől függő citosztatikus hatás jellemzésében, a DNS károsodás és a gyógyszermetabolizmus szerepének felderítésében Jeney András végzett úttörő munkásságot.
Nagyon sajnálatos, hogy a hatékonyságában sokszoros klinikai eredmények alapján bizonyított szerek gyártását a Chinoin beszüntette, és ezzel ezeknek a tudományos eredményekre alapozott (nem a naivan feltalált "csodaszerek" kategóriájába tartozó) gyógyszereknek a használata megszűnt, a hazánkban második halálozási okként szereplő rákos megbetegedések kezelésében.
A cukoralkohol származékok farmakológiai szempontból is igen fontos szerkezeti elemei az alkilező molekularészekhez különböző helyzetben és térhelyzetben levő hidroxil csoportok. Ez az egyéb biológiai alkilező szerektől eltérő szerkezeti tulajdonság a következő speciális, a tumorspecificitásra is kiható tényezőkben jelentkezik.
* Szomszédcsoport közreműködés révén elősegíti a DNS-el és egyéb nukleofilekkel végbemenő átalakulásokat (bioaktivációk)
* Az OH csoportok térhelyzetüktől függően befolyásolják a DNS keresztkötés lehetőségét
* Pozíciójuktól függően, intramolekulás reakciók révén DNS alkilezésre képtelen vegyületek létrehozásában vesznek részt (bioinaktiváció)
* Megváltoztatják farmakokinetikai és sejtmembrán penetrációs tulajdonságokat
* Igen sokfajta prodrog szintézisét teszik lehetővé
A daganatellenes hatás alapját képező DNS keresztkötéses reakciót, a szolvolitikus kémiai átalakulásokat és hatékony prodrogok szintézisének lehetőségét a szerző valamint Elekes Ilona és munkatársaik részletesen tanulmányozták az MTA Központi Kémiai Kutató Intézetében. Bebizonyították, hogy a keresztkötések ebben az esetben is elsősorban a két szálon lévő guanin bázisok N7 atomjai között jönnek létre. A reakció sebessége lényegesen függ az OH csoportok konfigurációjától. A szubsztituensek térkitöltését megnövelve (például az OH csoportok acilezésével) az eltérések igen jelentőssé válnak. A konfigurációtól függő reaktivitásbeli különbségeket a szubsztituensek és DNS között fellépő szterikus kölcsönhatásokkal értelmezték 1975-ben. A reakcióképesség magyarázatát a cukoralkohol származékoktól eltérő szerkezetű (például kénmustár) bifunkciós biológiai alkilező szerekre is érvényesnek találták.
Az eredmények nemcsak a konfigurációtól függő keresztkötéses reaktivitást magyarázzák, hanem tetszőleges számú szelektív prodrog tervezésére is iránymutatásul szolgálnak. Két szer gyógyszerré fejlesztését a Chinoin elkezdte, majd a sikeres preklinikai vizsgálatok után az új tulajdonos a klinikai vizsgálatoktól eltekintett.
A keresztkötések in vivo keletkezését, valamint a mono alkilezési és a bisguanil termékek arányát Institóris Etel (1981) igazolta és határozta meg. Kísérleteivel először bizonyította a vegyületek hatásmechanizmusát élő szervezetben végbemenő folyamatokban.
DNS két szála között nemcsak az alkilezőszerek, hanem a számos egyéb ágens is képes keresztkötést létrehozni. Ilyen a daganat kemoterápiában egyik legnagyobb volumenben alkalmazott vegyület: a cisplatin (14.). A keresztkötések a guanin N7 atomok között úgy jönnek létre, hogy a klóratomok Cl- ionok formájában leszakadnak, és helyükre kötődik be a nukleofil nitrogénatom. A részletes analízis kiderítette, hogy keresztkötések nem csak a két szál között, hanem az azonos szálon belül is keletkezhetnek. Ezek mennyisége a guanin részek DNS-en belül elfoglalt pozíciójától függően meghaladhatja a két szálas összekötődést. Mindkét típus gátolja a DNS funkciók kifejeződését, de a két szál közötti keresztkötés hatékonyabb és szelektívebb. A kedvező farmakológiai tulajdonságok növelése érdekében a platina vegyületek számos képviselőjét vizsgálták. Közülük gyógyászatban a carboplatin (15.) és oxaliplatin (16.) nyert legkiterjedtebb alkalmazást.
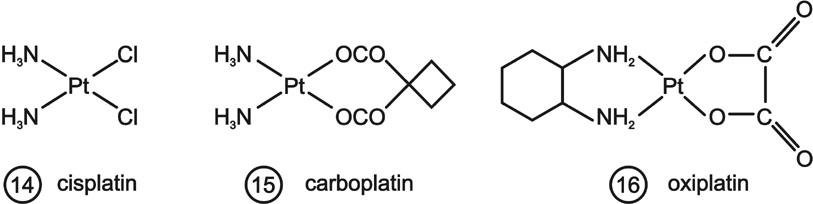
5. ábra Daganatterápiában használt platina vegyületek
A DNS-el interkalációs komplexet képező szerek
A DNS információk kifejeződését nemcsak a kovalens kötést létrehozó vegyületek, hanem azok az ágensek is előidézhetik, melyek a kettős hélix bázis párjai közé beékelődve azokkal komplexet hoznak létre. Ez az egészséges sejtek esetén nem kívánatos reakció a kovalens képzőkhöz hasonlóan felhasználható gyógyszeralkalmazási célból, amennyiben a korábban hangsúlyozott megfelelő szelektivitás biztosítható.
A bifunkciós alkilező szerekhez hasonlóan az interkalációs gyógyhatású anyagok megismerése is empirikus úton kezdődött el. Megfigyelték ugyanis, hogy egyes kondenzált aromás gyűrűt tartalmazó vegyületek (antibiotikumok), mint például az actinomycin D (17.), jelentős sejtproliferáció gátlást mutatnak. A DNS és említett vegyületek közötti kölcsönhatások tanulmányozása vezetett ahhoz a felismeréshez, hogy az anyagok interkalációs adduktokat hoznak létre a nukleinsavval. A vegyületek nagyobb százalékban a DNS kis árkában levő bázisok közé ékelődnek be. Az interkalációs komplex a DNS struktúra olyan torzulását hozza létre, hogy annak átírása lehetetlenné válik. A vegyületek ezen túlmenően egyéb DNS károsító hatással is rendelkeznek. Ezek egy része fokozza a proliferáció gátlást, ugyanakkor toxikus mellékhatásokat is okozhat. A széles körben alkalmazott antraciklinek, például a doxorubicin (18.) oxigén szabad gyököket generálnak, és ez jelentős kardiotoxicitást is okoz.
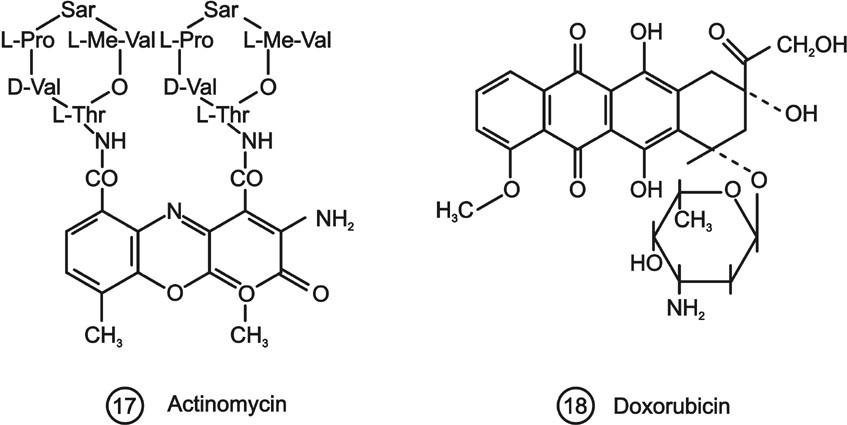
6. ábra DNS-el interkalációs komplexet képező gyógyszerek
Az interkalációs komplexképzők igen jelentős daganatellenes gyógyszercsoportot alkotnak, és jelenleg is széles körű kutatás tárgyát képezik. Mellékhatásaikat különböző vegyületekkel történő együttadással igyekeznek kiküszöbölni.
Magyarországon Hudecz Ferenc és munkacsoportja értek el igen jelentős eredményeket az interkalációs komplexképzők és a DNS közötti kölcsönhatások elméleti értelmezésében.
DNS-be beépülő nukleozid analógok
A vírus ellenes anyagok igen nagy, a tumor ellenes szerek jelentős részét nukleozid analógok képezik. A vegyületek többlépéses átalakulás (7. ábra) során nukleozid trifoszfátokká alakulnak, és polimeráz katalizálta reakciók révén épülnek be a DNS-be.
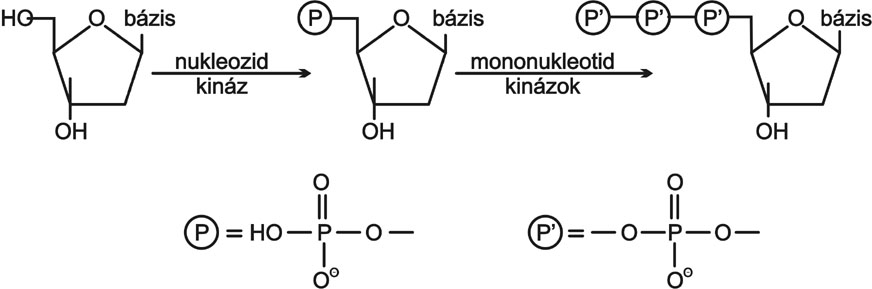
7. ábra Nukleozid-5'-trifoszfátok bioszintézise
A nukleozid analógok valójában prodrogok, melyek enzimes folyamatok útján bioaktiválódnak. A trifoszfátok közvetlen alkalmazása azért nem lehetséges, mert ezek erős negatív töltésük révén képtelenek a sejtmembránokon történő átjutásra. Az enzimatikus lépések sorozata ezen túlmenően a vírusellenes szelektív hatékonyságban vagy a specifikus antitumor hatásban is szerephez jut.
A nukleotid analógok polimerázok katalizálta DNS-be történő beépülésével olyan hibás szerkezetű nukleinsav keletkezik, mely információátadásra képtelen - megakadályozva mind a replikációs, mind a transzkripciós folyamatokat. A vírusterápiában alkalmazott vegyületek (8. ábra) jelentős része ezt úgy éri el, hogy nem tartalmaz a polimerizáció folytatásához szükséges 3'-OH csoportot (19.,20.,21.). Az ilyen szerkezetű nukleozid analógból keletkező trifoszfát nukleotid egysége a DNS-be beépülve annak terminálását okozza, ezzel megszakítva a DNS további bioszintézisét. (Összefoglalás: DeClercq, 2002.)
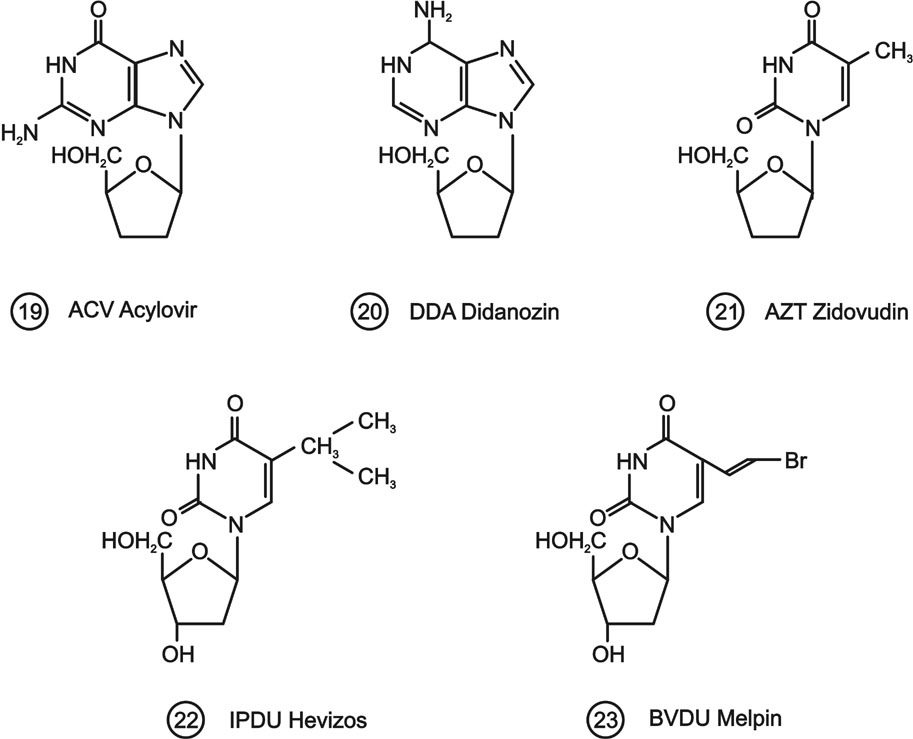
8. ábra Vírus ellenes nukleozid analógok
Más típust képvisel az MTA KKKI-ban kifejlesztett és a Biogal által gyártott Hevizos (22.). A vegyület 3'-OH csoportja révén képes a DNS lánc folytatására. Bizonyos mértékig be is épül a vírus DNS-be, de olyan szerkezet jön létre, mely enzimatikusan könnyen hasíthatóvá válik. Az átalakulás specificitása további szelektivitási tényező.
Az igen nagy HSV vírusspecificitású 5-bromvinil-dU-nak (23.) a DNS transzkripció mechanizmusára gyakorolt hatása még ismeretlen.
A fentiekben tárgyalt DNS-be való beépülés vagy a polimeráz működés gátlása a DNS információ átadás inhibiálását magyarázza. A szelektivitás az enzimek szubsztrátfüggő specificitásának a függvénye. A HSV ellenes anyagok esetén a 5'-monofoszfátok keletkezésére nagyfokú vírus indukálta enzimspecificitásban, következésképp vírus ellenes szelektivitásban jelentkezik A polimerázok működése is többé-kevésbé specifikus. Az 5-alkil-pirimidin analógok esetén (például Hevizos, 5-hexil-dU) a szubsztrát specificitás egészen nagyfokú is lehet, mint azt az MTA KKKI-ban végzett széleskörű vizsgálatok (Ötvös et al.,1987) bebizonyították.
A nukleozid analógok nemcsak a vírus, hanem a daganatterápiában is alkalmazást nyertek. Legjelentősebb ilyen származék az 5-fluor-2'-dezoxiuridin (24.). A vegyület gyors in vivo metabolizmusa miatt a gyógyászatban vagy különböző prodrogjait, vagy, gyakrabban a nukleobázis 5-fluor-uracilt (25.) alkalmazzák. Az FU-ból a szervezetben keletkezik az FdU enzimes reakcióban.
Az FdU többféle farmakológiai hatással bír. Legjelentősebb, hogy a 2'-dezoxi-uridin-5'-foszfát metilezésének gátlásával (timidilátszintáz inhibíció) gátolja a timidin-5'-foszfát keletkezését. Igen jelentős hatás az is, hogy a DNS-be illetve RNS-be beépülve megakadályozza azok funkciójának kifejlődését. Az igen kedvező hatékonyságú FdU illetve FU alkalmazhatóságát rontja a(z)
* jelentős, a nem megfelelő specificitásból eredő toxicitás
* in vivo bomlékonyság, mely az orális alkalmazást kizárja
* kialakult és fokozódó rezisztencia.
A hátrányok kiküszöbölésére illetve igen jelentős csökkentésére magyar kutatók - Kralovánszky Judit és Jeney András vezetésével - széleskörű vizsgálatokat végeztek 5-etil-2'-dezoxi-uridin modulátor alkalmazásával (Kralovánszky, 1999). Ezzel a módszerrel új, igen hatékony gyógyszerkombinációt hoztak létre.
A farmakológiában általánosan antimetabolitként említett nukleozid analógok közül kiemelendő a citozin-arabinozid (26) is, mely Magyarországon Alexan illetve Cytosar néven ismert.
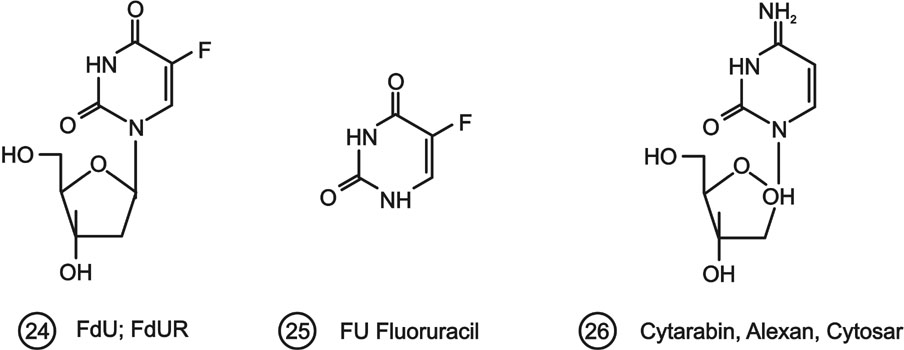
9. ábra Daganatellenes nukleobázis és nukleozid analógok
Antiszensz oligonukleotidok
Paul C. Zamecnik és Mary L. Stephenson amerikai kutatók 1978-ban publikálták, hogy bizonyos vírusok szaporodását oligodezoxinukleotidok gátolják mRNS-el képzett komplex-képzés (hibridizáció) útján. Ez a felismerés hatalmas lehetőséget nyitott meg az új gyógyszerek kifejlesztése előtt. Alapvető előnyük a szelektivitásban jelentkezik, melynek alapjait korábban már említettük. 15-25 tagszámú nukleinsav szakasz működése a megfelelő komplementer olionukleotiddal hibridet képezve olyan specificitást hoz létre, mely lehetővé teszi egyetlen, egy adott kóreseményben szerepet játszó fehérje keletkezésének inhibiálását. A vegyületeket természetes analógjaik alapján antiszensz oligonukleotidoknak nevezték el.
A természetes nukleotidokból felépített oligomerek az említett célból nem használhatók az élő szervezetben bekövetkező igen gyors lebomlásuk miatt. Gyógyszercélú felhasználásra az ezt kizáró módosított szerkezetű oligomerek szintézisére volt szükség. A világszerte igen nagy intenzitással megindult kutatások eredményeként ma ár több, mint ezerötszáz módosítás vált ismertté (több, mint ötven magyar eredetű). A módosítások első típusát a foszfátdiészter kötés tiofoszfátra való cseréje képviselte. Azóta az oligonukleotid részek minden egységét (bázisok, dezoxiribóz, internukleotid kötések) igen különböző szerkezeti elemekkel módosították.
Ezen alapvető követelménnyel együtt az antiszensz oligonukleotidoknak az alábbi tulajdonságokkal kell rendelkezniük:
* Stabil és specifikus hibridképzés a megfelelő mRNS-el
* Nukleáz rezisztencia
* Megfelelő sejtbejutási készség
* RNázH aktivitás
A specifikus hibridizációról és nukleáz rezisztenciáról történt említés. A sejtbejutási készség általános gyógyszerhatékonysági tényező. Az RNázH működés lehetővé teszi, hogy a szerek ne csak egy molekula mRNS transzlációját akadályozzák meg, hanem az inhibiált nukleinsav elbontása után, az antiszensz vegyület újabb mRNS-hez kötődjenek. A folyamat többször megismétlődhet. Az RNázH indukció nagymértékben függ az oligonukleotid módosítástól.
A kutatások a megfelelő biokémiai alapok birtokában - a 90-es évek közepén felmerült több ellenvetés dacára - a vártnál is gyorsabban vezettek új gyógyszerekhez. Közülük két vegyület - egy vírus ellenes és egy daganat ellenes szer - forgalomba is került. 1997-ben hét, 2000-ben tizennyolc, 2002-ben harminc anyag volt a klinikai vizsgálatok különböző fázisában (Hogrefe, 1999; 2002).
A farmaklógiai követelményeken kívül a széleskörű gyógyászati alkalmazáshoz szükséges volt olyan oligonukleotid gyártástechnológia kidolgozására, mely lehetővé tette a szerek megfizethető áron való hozzáférhetőségét. A hatalmas energiával és költséggel végzett fejlesztésekkel a problémát megoldottnak lehet tekinteni. A fejlődést az 1. táblázatban szereplő adatok szemléltetik.
1. táblázat
Hazai kutatók az antiszensz elv megismerése után igen rövid időn belül bekapcsolódtak a vizsgálatokba. Ehhez jó alapot adtak a hetvenes években elért szintetikuskémiai és polinukleotid bioorganikus kutatási eredmények (Ötvös, 1998). Éppen tíz éve, az MTA közgyűlésén a szerzőnek módja volt egy, az akkori DOTE Mikrobiológiai Intézetében (Gergely Lajos, D. Tóth Ferenc) HIV ellen in vitro rendkívül hatékonynak talált antiszensz oligonukleotid (KKKI-538) ismertetésére. Az OMFB, az OTKA és egy külföldi szponzor segítségével a vegyület sikeresen jutott túl a preklinikai vizsgálatokon, beleértve kutyákon végzett toxikológiai kísérleteket is. Az in vivo hatás bizonyítására és az ezt követő klinikai vizsgálatokra a megfelelő anyagmennyiség (minimálisan 1000 g) hiányában (előállítási költsége 1993-as áron több százmillió forint) sajnos nem kerülhetett sor.
A kilencvenes években a DEOEC Biológiai Intézetben létesült egy antiszensz oligonukleotid kutatórészleg. 2001-ben közölt (Szatmári - Aradi, 2001) alapkutatási eredményeikre támaszkodva Aradi János munkacsoportja olyan telomeráz inhibitorokat fejlesztett ki, melyek a Watson-Crick bázispárosodás szabályai szerint kölcsönhatásba lépnek a telomeráz RNS templát régiójával, majd egy kapcsolt kémiailag módosított oligonukleotid szakasz segítségével a protein alegységhez kötődnek. A vegyületek igen nagyfokú telomeráz gátló hatást fejtenek ki. Ezen az alapon potenciális tumor terápiás ágenseket sikerült megismerni.
Az MTA Kémiai Kutatóközpontban a szerző irányításával három éve indultak meg kutatások egy új oligonukleotid gyógyszermegismerési koncepció alapján. Az antiszensz irányított prodrog terápiának (ADPT - antisense directed prodrug therapy) elnevezett módszer egyesíti a prodrog és antiszensz szelektivitási elv előnyeit. Ezzel elméletileg szuperszelektív szerek előállítása várható.
Az új elv azon az igen egyszerű elgondoláson alapszik, hogy olyan prodrogokat állítsunk elő, melyek egyik fele antiszensz molekula, a másik hozzá kovalensen kapcsolt hatékony gyógyszer. Az antiszensz rész felismeri a hibás nukleinsav adott szakaszát, majd a drog rész enzim közreműködéssel felszabadul. Utóbbit modellkísérletek igazolták (Ötvös, 2002).
Ezzel a mechanizmussal lokálisan és szelektíven nagy gyógyszer- koncentráció érhető el, ami a nagymértékben megnövelt hatékonyságban jelentkezik. Ehhez adódik hozzá az antiszensz oligonukleotid saját hatása. A gyógyszertervezésben igen nagy előny, hogy mind az oligomer, mind a kapcsolt egység ismert és kipróbált gyógyszer lehet. A variálási lehetőség a már eddig ismert szerek figyelembevételével is igen nagyszámú.
Jeney András vezetésével végzett vizsgálatokban a módszert több oligonukleotid-FdU konjugátummal tanulmányozták. A vegyületek az FdU különböző rákos sejtek szaporodásgátló hatását 5-105-szörös (!) mértékben növelték meg. Az FdU konjugátumok esetén külön előnyt jelent, hogy egy oligonukleotid 3'-végéhez, a dezoxinukleozid bifunkciós jellegéből adódóan, több FdU molekula is kapcsolható.
Nem kell hangsúlyozni, hogy milyen gyógyászati jelentősége volna az ADPT-nek, ha a hatékonyságnövelés in vivo is bizonyítást nyerne. Remélhetőleg sikerül olyan financiális lehetőségekhez jutni, hogy az ilyen típusú vizsgálatok elkezdődjenek, és erre alapozva a vegyületek különböző (elsősorban daganatos és vírusfertőzés okozta) betegségek leküzdésében alkalmazási lehetőséghez jussanak.
IRODALOM
- Brookes, Peter - Lawley, Philip D. (1961): Reaction of Mono- and Difunctional Alkylating Agents with Nucleic Acids. Biochemical Journal. 80, 496-503.
- De Clercq, Erik (2002): Strategies in the Design of Antiviral Drugs. Nat. Rev. Drug Disc. 1, 13-25.
- Borvendég János (főszerk.): Gyógyszer Kompendium 2002. Az Országos Gyógyszerészeti Intézet hivatalos kiadványa. MediMédia Információs Kft., Budapest
- Jacobson, Leon Orris - Spurr, C. L. - Barron, E. S. G. - Smith, T. - Lushbaugh, C. - Dick G. F. (1946): Nitrogen Mustard Therapy. Studies on the Effect of Methylbis(2-chloroethyl)amine Hydrochloride on Neoplastic Diseases and Allied Disorders of the Hemopoietic System. The Journal of the American Medical Association. 134, 263-271
- Jeney András (2001): in Fürst Zsuzsanna (szerk.). Farmakológia. Medicina, Budapest, 1066.
- Haddow, A. (1949). Mode of Action of the Nitrogen Mustards - A New Working Hypothesis and its Possible Relation to Carcinogenesis. Proceedings of the National Cancer Conference 1949. 88-94
- Hogrefe, R.I. (1999): An Antisense Oligonucleotide Primer. Antisense and Nucleic Acid Drug Development. 9, 351-357. Table 1 updated 2002. Web: www.trilinkbiotech.com
- Hudecz Ferenc - Kajtár Judit - Szekerke Mária (1981): Interaction of the Antitumour Drug 4'-(9-acridinylamino)-N-methane-sulfon-m-anisidine.HCl (mAMSA) with Nucleic Acids. Nucleic Acid Research. 9, 24. 6959-6973.
- Institóris Etel (1981): In Vivo Study on Alkylation Site in DNA by the Bifunctional Dianhydrogalactitol. Chemico-Biological Interactions. 35, 2, 207-216.
- Institóris László - Dzurillay E. - Sebestyén J. H. - Jeney A. - Pethes Gy. (1971): Correlation of the Differences of Biological Effects with Distribution Parameters of 1,6-dibromohexitols.(in German). Zeitschrift fur Krebsforschung. 75, 2, 133-145.
- Kopper László (2002) in Kopper László - Jeney András (szerk.): Onkológia a géntől a betegágyig. Medicina, Budapest, 364.
- Kralovanszky Judit - Katona Cs. - Jeney A. - Pandi E. - Noordhuis, P. - Erdelyi-Tóth V. - Ötvös L. - Kovács P. - Clasina, L. - Vand der Wilt, C. - Peters, G. J. (1999). 5-Ethyl-2'-deoxyuridine, a Modulator of Both Antitumour Action and Pharmacokinetics of 5-fluorouracil. Journal of Cancer Research and Clinical Oncology. 125, 12, 675-684.
- Ötvös László - Elekes I. (1975): Steric Hindrance in the Reactions of DNA with Bifunctional Alkylating Agents. Definition of "t" and "m" Substituents. Tetrahedron Letters. 29, 2477-2480.
- Ötvös László - Sági J. - Kovács T. - Walker, R.T. (1987): Substrate Specificity of DNA Polymerases. I. Enzyme-catalysed Incorporation of 5-(I-alkenyl)-2'-deoxyuridines into DNA. Nucleic Acids Research 15, 4, 1763-1777.
- Ötvös László (1998): Nucleosides and Nucleotides Containing 5-Alkyl Pyrimidines in Chapleur, Yves (Ed.): Carbohydrate Mimics. Chemistry and Molecular Pharmacology. Wiley-VHC Press, NY. 537-552.
- Ötvös László - Bajor Z. - Kraicsovits F. - Sági Gy. - Tegyey Zs. (2002): Synthesis and Enzymatic Characterization of P1-thio-P2-oxo Trideoxynucleoside Diphosphates Having AZT, FdU, or dT at the 3'-position. Nucleosides, Nucleotides & Nucleic Acids. 21, 79-92.
- Süli-Vargha Helga - Jeney, A. - Lapis, K. - Medzihradszky, K. (1987):
Synthesis and Antitumor Activity of N-terminal Proline Containing Peptide-(chloroethyl)nitrosoureas. Journal of Medicinal Chemistry. 30, 583-586.
- Szatmári István - Aradi János (2001): Telomeric Repeat Amplification, without Shortening or Lengthening of the Telomerase Products: A Method to Analyse the Processivity of Telomerase Enzyme. Nucleic Acids Research. 29, 2, e3
- Zamecnik, Paul C. - Stephenson, Mary L. (1978): Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proceedings of the National Academy of Sciences of the USA. 75, 285-288.
Kulcsszavak: DNS, RNS, gyógyszer, kölcsönhatás, célpont, daganat terápia, antivirális hatás
Év Vállalat Ár ($/g) 1990 ISIS 42000 1997 ISIS 1000 2002 (becslés) ISIS ~100 Amersham 2002 (becslés) Poligo ~50 Avecia
1. táblázat Oligonukleotidok előállítási költségeinek alakulása