50 éves a "kettős csavar"
Penke Botond
az MTA levelező tagja
Datki Zsolt
PhD hallgató
Zarándi Márta
a kémiai tudomány kandidátusa
SZTE Orvosi Vegytani Intézet és MTA Fehérjekémiai Kutatócsoport, Szeged
NEURODEGENERATÍV BETEGSÉGEK KÉMIAI ÉS BIOKÉMIAI HÁTTERE
1. Bevezetés
A neurodegeneratív betegségek nagy része fehérje aggregátumok képződésével
kezdődik. Az aggregátumok részben a sejteken kívül helyezkednek el (ilyenek
például az Alzheimer-plakkok), részben viszont a sejten belül zárványokat
alkotnak (például alfa-synuclein a Lewy-testekben, hiperfoszforilezett tau-fehérje
neurofibrilláris kötegekben). A legismertebb neurodegeneratív betegségeket és a
jellemző fehérje aggregátumokat az 1. táblázat foglalja össze.
Betegség Fehérje aggregátum
1. Alzheimer-kór béta-amiloid (plakkok) tau (neurofibrilláris kötegek)
2. Parkinson-kór alfa-synuclein / ubiquitin
3. Lewy-testes demencia alfa-synuclein
4. FTDP-17 tau (Pick-testek)
5. Huntington-kór poli-glutamin / ubiquitin
6. Prion betegség (Creutzfeld-Jacob-kór) prion protein
7. Pick-betegség tau (Pick-testek)
8. Amiotróf lateral sclerosis ubiquitin
1. Táblázat A neurodegeneratív betegségek közös mechanizmusa: fehérje
aggregátumok képződése
Egy és ugyanaz a fehérje aggregátum többféle betegségben is megjelenhet. A
tauopathiák nagy családja az Alzheimer-kóron kívül a Pick-betegséget valamint a
frontotemporális demenciát és parkinsonizmust (FTDP-17) is magába foglalja, az
alfa-synuclein fibrillumok egyaránt megjelennek a Parkinson-kórban és a
Lewy-testes demenciában. Sokszor nem könnyű a neurodegeneratív betegségeket
egymástól elkülöníteni, éppen a közös fehérje aggregátumok miatt. Nagyon
jellemző viszont az idegsejtek károsodásának, pusztulásának a helye, így a
legtöbb neurodegeneratív betegség régió- sőt sejtspecifikus, legalábbis a
betegség korai szakaszában. A kórképek közös mechanizmusára utal az a tény,
hogy a felsorolt fehérjék valamennyi esetben konformációváltozást szenvednek
(alfa-hélix -> béta-szalag átalakulás) és béta-szerkezetű polipeptid láncokat tartalmazó
szálakká, fibrillumokká alakulnak át (Mager, 2002). A fibrillumok, sőt a
kisebb, diffúzibilis aggregátumok is enzimrezisztensek és neurotoxikus
hatásúak. A szakirodalom megegyezik abban, hogy a felsorolt neurodegenerációs
betegségek végső oka bizonyos fehérjék nem megfelelő feltekeredése,
gombolyodása (misfolding), a hibás fehérje térszerkezetek kialakulása.
2. A neurodegeneratív betegségek genetikai háttere
Nem minden esetben tisztázott a neurodegeneratív betegségek genetikai háttere.
A 2. és 3. táblázat röviden összefoglalja a legfontosabb betegségek öröklődését
és az eddig ismert fontosabb mutáns géneket, genetikai faktorokat.
Betegség Öröklődés, genetikai háttér
1. Alzheimer-kór Sporadikus/Autoszom. domináns
2. Parkinson-kór Sporadikus/Autoszom. recesszív
3. Lewy-testes demencia Sporadikus/Autoszom. recesszív
4. FTDP-17 Autoszom. domináns
5. Huntington-kór Autoszom. domináns
6. Prion betegség (részben öröklött)
7. Pick-betegség Sporadikus
8. Amiotróf lateral sclerosis Sporadikus/Autoszom. domináns
2. Táblázat: A neurodegeneratív betegségek genetikai háttere 1.
Betegség Mutáns Gének
1. Alzheimer-kór APP, PS-1, PS-2, apoE epszilon4, TAU, GSK-3béta, ChAT, MTHFR
2. Parkinson-kór tau, N-acetil-transzferáz, PANK2, MTDN 1-2
3. Huntington-kór (CAG ismétlődés)
4. Amiotróf lateral sclerosis SOD1, SMN, tau
5. Down-kór tau, ChAT
3. Táblázat: A neurodegeneratív betegségek genetikai háttere 2.
A tisztán öröklött génhibán alapuló neurodegenerációs kórkép viszonylag ritka,
ilyen például a Huntigton-kór, amelynél egy poliglutamin-peptidlánc (egy CAG
triplett ismétlődése miatt fellépő rendellenesség) aggregációja váltja ki az
idegsejtek halálát. Ismerünk egy sorozat pontmutációt az alfa-synuclein génben,
amelyek kiváltják a familiáris Parkinson-kórt illetve a Lewy-testes demenciát.
A neuronok mikrotubuláris rendszeréhez kapcsolódó tau-fehérjék génjének
pontmutációi abnormális hiperfoszforilációt idéznek elő, emiatt a
mikrotubuláris rendszer széthullik, s ez nagymértékben hozzájárul a sejt
halálához. Az igen intenzív kutatómunka ellenére még sok esetben nem tisztázott
a neurodegeneratív betegségek genetikai háttere.
Az Alzheimer-kór a leggyakoribb demenciás kórkép, a memóriavesztéses
betegségeknek több mint 50%-áért felelős. A korai jelentkezésű (a 40-60. életév
között fellépő) familiáris Alzheimer-demencia (AD) előfordulása 5% alatt van.
Az öröklődő betegségért néhány fehérje (például amiloid prekurzor protein,
presenilin-1 és 2, tau-fehérje) mutációi a felelősek, ezek a mutációk jól
ismertek, de igen ritkák. A mutációk az 1, 12, 14, 19 és 21. kromoszómában
lépnek fel (4. táblázat). Az Alzheimer-kór genetikai alapjáról Cacabelos írt
részletes összefoglalót (1999).
Kromoszóma Gén Jelentőség Korkezdet
1 presenilin-2 (PS2) Korai familiáris AD, 50-70
1q 31-42 autoszom. domináns
12 alfa2-makroglobulin LRP1 késői AD >75
14 presenilin-1(PS1) korai familiáris AD >40
14q 24.3, c-FOS, autoszom.domináns
14q 24.3-31
19 APO-E epszilon4 késői familiáris AD >55
19q 12-23.3
21 amiloid prekurzor protein korai familiáris AD, >50
21q 11.1-21.1 autoszom. domináns
4. Táblázat: Az Alzheimer-kór genetikája: familiáris formák
A viszonylag ritka mutációk mellett az Alzheimer-kór gyakori és komoly
rizikófaktora az apo-E fehérje polimorfizmusa (Tariska, 2000). Ez a fehérje 299
aminosavból áll, főleg a lipidek transzportjáért és anyagcseréjéért felelős.
Három különböző allélje van (epszilon2, epszilon3 és epszilon4), amelyek csak egy-egy pontmutációban
különböznek egymástól, így egy vagy két aminosav különbséget találunk a
polipeptid láncban (5. táblázat). Az epszilon2 allélnek megfelelő fehérje (Cys 112,
Cys 158) inkább védő hatású, viszont az epszilon4 allélok (Arg 112, Arg 158) jelenléte
tizenhétszeresére növeli az Alzheimer-kór fellépésének gyakoriságát. Az eddigi
vizsgálatok szerint az AD-esetek 15-20%-áért az apo-E epszilon4 allél jelenléte a
felelős.
112. hely 158. hely
APOE allél Triplet Aminosav Triplet Aminosav
epszilon2 TGC Cys TGC Cys
epszilon3 TGC Cys CGC Arg
epszilon4 CGC Arg CGC Arg
5. Táblázat: Az apo-E gén polimorfizmusa mint az Alzheimer-kór kiváltó oka
Ha valamennyi ma ismert genetikai faktort összeadunk, azt találjuk, hogy az
Alzheimer-demenciák 20-25%-át genetikai tényezők váltják ki. A késői kezdésű
(65. életév után jelentkező) AD-k legnagyobb részénél nem ismerjük a kiváló
okokat. A betegség többtényezős, multifaktoriális eredetűnek látszik. A
neurotoxikus béta-amiloidok szintéziséért, illetve lebontásáért felelős proteázok
egyensúlyának zavara, a szabad gyökök megkötéséért felelős enzimek
alulműködése, az idegsejtekben folyó ATP-termelés csökkenése, a vér-agy gát
permeábilitás megnövekedése egyaránt hozzájárulhatnak a betegség
kialakulásához. Magyarországon az Alzheimer-kór különösen gyakran társul
vaszkuláris eredetű demenciával; az agyi erek szklerózisa az Alzheimer-kór
egyik rizikófaktora. Bizonyos vaszkuláris faktorok változása (például magas
vérnyomás, az agyi kapillárisok állapota, stb.) fontos kóroki tényező lehet. A
legújabb kutatások kiderítették, hogy az Alzheimer-kór kialakulása igen hosszú
folyamat (15-20 év), emiatt igen valószínű, hogy a középkorú populáció (40-55.
év) kezeletlen magas vérnyomásos betegeinél nagymértékben megemelkedik az AD
kockázata. Komoly kockázati tényező az életkor előrehaladása, az elszenvedett
koponyatraumák és a tartós oxigénhiány.
3. A leggyakoribb neurodegenerációs betegség, az Alzheimer-kór
morfopatológiája, biokémiája és kialakulásának mechanizmusa
Az Alzheimer-demenciát Alois Alzheimer már 1907-ben leírta, és a következő
jellemzőit sorolta fel: az agyszövet nagyfokú atrófiája; amiloid plakkok
kialakulása bizonyos agyterületeken, illetve neurofibrilláris kötegek
megjelenése. A betegség elsősorban a szinapszisok és a kolinerg neuronok
pusztulásával jár (Selkoe, 1991). Feltűnő a neuronok mitokondriumainak
sérülése, elfajulása. Ez komoly ATP-hiányt okoz, többen ezt tartják az idegsejt
pusztulás végső okának. Az igazi okokat azonban jóval korábbi lépéseknél kell
keresni.
Nagyon sokan vizsgálták a (csak mikroszkóppal látható) amiloid plakkok
szerkezetét és összetételét. A betegség előrehaladásával a plakkok is
változnak: a kezdetben diffúz plakk több lépésben szenilis plakká alakul,
közepén "keményítőszerűen" festődő maggal (innen jön az amiloid név). A mag
kissé szivacsos állományú és főleg béta-amiloid peptideket, tau-fehérjét,
lipofuscint és más anyagokat tartalmaz. Bizonyos festékek (például Kongóvörös,
tioflavin) specifikusan kötődnek a béta-amiloidokhoz, ennek az a magyarázata, hogy
a plakkokban a polipeptidek ún. béta-redőzött réteg vagy béta-szalag szerkezetet
vesznek fel. Ezek egymáshoz kapcsolódnak, aggregálódnak és hosszú fibrillumokat
alakítanak ki. Az amiloid aggregátumok neurotoxikus hatásúak: a plakk közelében
húzódó, a plakk magjával érintkező axonok degenerációját indítják el. (A
sejttest sokkal kevésbé érzékeny a béta-amiloid aggregátumokra). A betegség
előrehaladásával, súlyosodásával szinte egyenes arányban nő az elhalt
idegsejtekből képződő neurofibrilláris kötegek mennyisége, ezeket főleg a már
említett "abnormálisan" foszforilezett (túl sok foszfátészter-csoportot
tartalmazó) tau-fehérjék alkotják. Az amiloid plakkok száma nem áll mindig
arányban a betegség súlyosságával.
Az Alzheimer-kór az öregkor betegsége, de ritka esetben fiatalabb korban is
jelentkezik. Évtizedes vita után el kell fogadnunk, hogy a betegséget a
béta-amiloid peptidek túltermelődése, aggregációja váltja ki, ez az indító lépés.
(Ez nem áll ellentétben azzal a ténnyel, hogy bizonyos tau-fehérje mutációk
béta-amiloid képződés nélkül is neurodegenerációhoz vezetnek a tau-fehérjék
hiperfoszforilezése révén. Ezt a betegséget a plakkok hiánya miatt nem
tekinthetjük Alzheimer-kórnak). A fiatalabb korban, a 40-65. életév között
jelentkező Alzheimer-kórt főleg az amiloid prekurzor protein (APP) és a
presenilinek mutációi idézik elő (4. táblázat): ezek hatására az APP-ből nagy
mennyiségű, igen könnyen aggregálódó neurotoxikus peptid képződik. Az APP egy
sejtadhéziós fehérje, a szinaptikus membránokban van jelen nagy
koncentrációban, pontos biológiai szerepét nem ismerjük. Különböző molekuláris
formái vannak: a neuronokban a 695 aminosavas, a glia sejtekben a 751 illetve
770 aminosavas forma fordul elő (Tanzi, 1988). A központi idegrendszert ért
traumák hatására az APP nagy mennyiségben szabadul fel. Állandóan ismétlődő
agyi traumák illetve hipoxia hatására sok APP termelődik, ez nagyfokú béta-amiloid
képződést okoz. Ezzel magyarázzák a boxolók dementia pugilisticaját, de a
gyakori hipoxiás állapotba kerülő sportolók (hegymászók illetve búvárok) korai
demenciáját is.
Az idegsejtek elhalásának pontos mechanizmusát Alzheimer-kór esetén még nem
sikerült teljesen igazolni. Sejtszinten, molekuláris szinten a következő
lépésekben képzeljük el a betegség kialakulását az amiloid-kaszkád hipotézis
alapján (Hardy, 1992).
1. Kisebb-nagyobb agyi traumák, hipoxia, esetleg genetikai faktorok APP
túltermelődéshez vezetnek, a prekurzorból a normálisnál nagyobb mennyiségű
béta-amiloid peptid képződik. Idősebb korban ezt elősegíti a lebontó proteázok
csökkent működése is.
2. A sejtek felszínén, az extracelluláris térben a béta-amiloid peptidek
neurotoxikus aggregátumokat képeznek (Lambert, 1998). Ezek különböző
nagyságúak, a kisméretű, diffúzibilis aggregátumoktól a hosszú szálakig sokféle
forma előfordulhat, de valamennyi forma toxikus. (Maguk a monomer béta-amiloidok
nem toxikusak.)
3. A béta-amiloid aggregátumok megkötődnek az idegsejtek membránfehérjéin.
Valószínű, hogy a béta-amiloid aggregátumnak klasszikus értelemben nincs
receptora, hanem többféle fehérjén meg tud kötődni. (Van olyan vélemény, hogy a
ß-amiloid aggregátum számára "minden membránfehérje kötőhely", de ezt a
kísérletek nem igazolják). A fehérjék egy része G-proteinnel kapcsolt receptor.
4. A béta-amiloid-membránfehérje kötődés hatására Ca2+ ionok áramlanak be a
sejtbe. Igazolt, hogy az amiloid peptidek megkötődnek az NMDA-receptoron, az
integrin-receptorok bizonyos típusain, az APP-n, a RAGE-receptoron, stb. Mivel
az amiloid aggregátum enzimrezisztens és tartósan ott marad a membránon, a
Ca2+-beáramlás állandósul.
5. A Ca2+-jel aktiválja a protein kinázokat (például Cdk5, GSK3béta), és
megkezdődik a mikrotubuláris-rendszert alkotó tau-fehérjék abnormális helyen
történő foszforilezése (hiperfoszforileződés). Eltolódik a
foszforiláz-foszfatáz enzimegyensúly, az abnormális tau-fehérjék nem képesek a
mikrotubulusok szervezésére, a szerkezet összeomlik. A hiperfoszforilezett
tau-fehérjék lassan neurofibrilláris kötegekké aggregálódnak.
6. A megemelkedett Ca2+-szint egyedül is elegendő a mitokondriumok
károsításához. A kettős membrán felszakad, a sejtlégzés és az ATP képződés
leáll, nagy mennyiségű szabad gyök képződik. A mitokondriumból kiszabaduló
faktorok (apoptózis indukáló faktor, citokróm-c) beindítják a neuron elhalását.
7. Az axonok is központi szerepet játszanak a neurodegenerációban. A
mikrotubuláris rendszer összeomlásával megszűnik az axonális transzport. A
neuron lassan elveszíti dendritjeit és axonját, legömbölyödik (dezarborizáció,
vezikularizáció) és lassan elhal.
4. Az Alzheimer-kór kezelése, a megelőzés lehetőségei
Az elhalt neuronokat már nem lehet visszahozni, viszont a patomechanizmus
ismeretében ma már nem reménytelen a betegség kezelése és a racionális
gyógyszertervezés.
A kolinerg rendszer részleges kiesését, az acetilkolin-szint csökkenését
kolinészteráz-gátlókkal próbálják kivédeni - de ez természetesen csak tüneti
kezelést jelent. Újabb kezelési lehetőség a Memantine (dimetil-Amantadin)
alkalmazása. Ez a gyógyszer bekötődik az NMDA-receptor ioncsatornájába, és
megakadályozza a Ca2+-beáramlást. A szakirodalom igen jó eredményekről számol
be: ha az idegsejtek még nem haltak el, csak "fojtogatja" őket az amiloid
aggregátum, a Ca2+-beáramlás megakadályozásával ezek a sejtek "felélednek",
visszanyerik működőképességüket, és Memantine hatására a betegek állapota javul.
A szakirodalom részletesen beszámol a reaktív szabad gyököket "eltakarító"
anyagok (C-vitamin, E-vitamin, flavonoidok) kedvező hatásáról is. Ezek mellett
a szteroid-hormonok szerepével, hatásmechanizmusával is sokan foglalkoznak.
Az Alzheimer-kutatás legújabb iránya abból indul ki, hogy a béta-amiloid peptidek
központi szerepet töltenek be a betegség kialakulásában, ezek keletkezését,
aggregációját illetve sejtmembránhoz való kapcsolódását kell megakadályozni.
A béta-amiloid peptidek képződésének gátlása
A prekurzor fehérje lebontásában három enzim játszik kulcsszerepet: az alfa-, béta-
és gamma-szekretáz. Ha az alfa-szekretáz hasít, az APP-ből vízoldható, nem toxikus
peptidek sokasága képződik. Mindig van lehetőség viszont az alternatív
hasításra: a béta-szekretáz, majd gamma-szekretáz működése olyan, 40-42 aminosavból
álló peptideket hasít ki az APP-ből, amelyek igen könnyen aggregálódnak, s
emiatt neurotoxikusak (béta-amiloidok). A béta-amiloid monomerek kis mennyiségben
mindig képződnek és neuromodulátor hatásúak: csökkentik a kolinerg-receptorok
ingerelhetőségét. Az alternatív hasítás csak akkor veszélyes, ha nagy
mennyiségű béta-amiloidot termel, és ez aggregálódik. Megfelelő enzimgátlókkal a
béta-amiloid képzés csökkenthető, és (elvileg) a kór előrehaladása
megakadályozható.
A béta-szekretáz egy aszpartil-proteáz (Vassar, 1999), Röntgen-diffrakciós
szerkezete ismert. Számos laboratóriumban (így nálunk is) folyik a specifikus
béta-szekretáz inhibitorok számítógépes tervezése és szintézise. Úgy tűnik, a
béta-szekretáz gátlása nem okoz különös mellékhatásokat kísérleti állatokon.
A gamma-szekretáz szintén aszpartil-proteáz, egy bonyolult membránfehérje-komplex.
Az az érdekessége, hogy a polipeptidláncot éppen a membrán belsejében középen
hasítja, az APP-transzmembrán régiójában. A gamma-szekretáz röntgendiffrakciós
szerkezete nem ismert, ennek ellenére számos inhibitorát ismerjük a
szakirodalomból.
A sejtmembrán lipid összetétele nagymértékben befolyásolja a béta- és
gamma-szekretázok aktivitását. Nagy mennyiségű koleszterin jelenléte a membránban
növeli a béta- és gamma-szekretáz aktivitását, így a koleszterin bioszintézist gátló
gyógyszerek (Lovastatin, Mevastatin, stb.) jó hatással lehetnek
Alzheimer-kórban. Ugyanakkor a többszörösen telítetlen omega-3 zsírsavak
(dokoza-hexaénsav, C22:6, DHA és eikozapentaénsav, C20:5, EPA) jelenléte a
membránban csökkenti a béta- és gamma-szekretáz aktivitást és a keletkező béta-amiloidok
mennyiségét. Intenzív kutatómunka folyik olyan diéta kidolgozására, amelyik
többszörösen telítetlen zsírsavak bevitelével akadályozza meg az Alzheimer-kór
kialakulását, illetve lassítja le a betegség előrehaladását.
A béta-amiloidok aggregációja és toxicitása
A béta-amiloidok aggregációja az Alzheimer-kór fontos rizikófaktora.
Megvizsgáltuk, hogyan függ össze az amiloid peptidek szerkezete, aggregációs
képessége és neurotoxicitása. Az aggregációt FT-IR spektroszkópiával követtük
nyomon, a neurotoxicitást differenciált SH-SY5Y neuroblasztóma tenyészeten
MTT-teszttel mértük. (A teszt a sejtek életképességét, redukciós potenciálját
méri, egy tetrazolfesték formazánná történő redukciós átalakulásával).
Megmértük a különböző lánchosszúságú ß-amiloid peptidek, valamint a csupán
D-aminosavat tartalmazó peptidek illetve a fordított (reverz) aminosav-sorrendű
analógok aggregációs készségét és toxicitását. Az eredményeket az 1. és a 2.
ábra mutatja.
Szekvencia Aggregáció Toxicitás
1. Abéta 1-40 +++ +++
2. Abéta 1-42 ++++ ++++
3. Abéta 25-35 +++ +++
4. Abéta 31-35 +++ +++
5. csupa D-Abéta 1-40 +++ +++
6. reverz Abéta (42-1) - -
7. reverz Abéta (35-25) - -
1. ábra: Amiloid peptidek aggregációja és toxicitása.
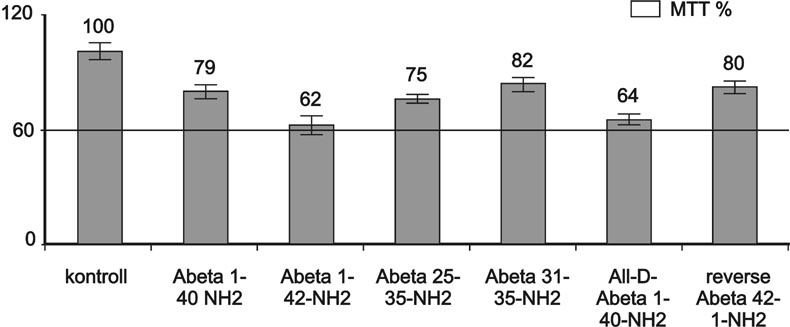
2. ábra: Abéta-peptidek toxicitása MTT-teszten.
Az Abéta-peptidek toxicitása jó korrelációban van az aggregációjukkal. Néhány kis
Abéta-fragmens is toxikus (25-35, 31-35). A csupán D-aminosavakból felépülő Abéta
1-40 is toxikus, mivel gyorsan aggregálódik. Ezzel szemben a fordított sorrendű
Abéta 42-1 nem képez aggregátumokat, és alacsony toxicitást mutat.
Toxikus amiloid aggregátumok képződésének megakadályozása
A béta-amiloid polipeptid láncához különböző típusú vegyületek kapcsolódhatnak
ionos kötéssel illetve másodlagos kötésekkel. Az ilyen vegyületek
megakadályozzák a peptidlánc aggregációját, és elvileg alkalmasak lehetnek az
Alzheimer-kór kezelésére. Ezeket az anyagokat összefoglaló néven
béta-szerkezetrombolóknak (béta-sheet-breaker) nevezzük. Legismertebb ezek közül az
azofesték jellegű Kongóvörös.
Tjernberg (1996), majd később Soto (1996) kutatócsoportja ismerte fel, hogy a
béta-amiloid 16-20 illetve 17-21. pentapeptid részlete megakadályozza az
aggregációt. Számos módosított béta-szerkezetromboló peptidet állítottak elő, ezek
közül a legismertebb a Leu-Pro-Phe-Phe-Asp pentapeptid.
Kutatócsoportunkban számítógépes molekulatervezéssel, a béta-amiloid peptidlánc
felszínére történő illesztéssel (dokkolás, AUTODOCK-program) kerestünk olyan
peptideket, amelyek viszonylag erős kötéssel illeszkednek az amiloidhoz, és
megakadályozzák az aggregációt. Ezeket szintetikusan is előállítottuk, majd in
vivo és in vitro tesztekben megvizsgáltuk a szintetikus peptidek neuroprotektív
hatását. A következő vegyületek bizonyultak legjobbnak:
Leu-Pro-Tyr-Phe-Asp
Arg-Val-Val-Ile-Ala
Ezek a vegyületek önmagukban is potenciális gyógyszerek. A vér-agy gáton
áthaladó, enzimrezisztens analógok és peptidomimetikumok tervezése és
szintézise szintén megkezdődött.
Az aggregált amiloid és a membránfehérjék közötti kötődést gátló neuroprotektív
anyagok: funkcionális antagonisták
Még évekkel ezelőtt azt találtuk, hogy a béta-amiloid egy rövid fragmense, az
Ile-Ile-Gly-Leu tetrapeptid-amid és származékai megakadályozzák a ß-amiloid
peptidek neurotoxikus hatásának kifejlődését (Laskay, 1997). Mivel ezek a
peptidek nem ß-szerkezetrombolók, inkább oly módon hatnak, hogy megakadályozzák
az aggregátumok kötődését a membránfehérjéken. Így lehetetlenné válik a
Ca2+-ionok beáramlása és nem jöhet létre apoptózis. A vizsgálatok igazolták ezt
az elképzelést, így erre a vegyületcsoportra egy új nevet kellett bevezetnünk:
ezek az ún. ASBIM (Amyloid Surface Binding Molecule) vegyületek. In vitro és in
vivo tesztekben a következő peptidek bizonyultak legjobbnak:
propionil-Ile-Ile-Gly-Leu-amid
Arg-Ile-Ile-Gly-Leu-amid
Phe-Arg-His-Asp-Ser-amid
Ezek a vegyületek a béta-amiloidok funkcionális antagonistáinak tekinthetők.
Valamennyi peptid tartalmaz béta-amiloid szekvencia részletet. Az utolsó két
vegyület ún. RGD analóg, tehát az integrin-fehérjékhez kötődve is kifejtheti
hatását. Ezek a vegyületek vezérvegyületként szolgálnak olyan enzimrezisztens,
a vér-agy gáton áthaladó peptidomimetikumok tervezéséhez és szintéziséhez,
amelyek az Alzheimer-kór igazi gyógyszerei lehetnek.
Összefoglalva: az Alzheimer-kór patomechanizmusának ismeretében új utak nyíltak
a gyógyszertervezés előtt: enzimgátlók, béta-szerkezetrombolók és a membránfehérje
- aggregált amiloid kölcsönhatást gátló vegyületek lehetnek a jövő potenciális
gyógyszerei. Valószínű, hogy a fehérje aggregátumok keletkezését és toxicitását
megakadályozó vegyületek alkalmazásának alapelve kiterjeszthető a többi
neurodegeneratív betegség megelőzésére is.
IRODALOM
- Cacabelos, Ramon (1999): Association of Genetic Risk Factors in Alzheimer's
Disease. In: Iqbal, Khalid (ed): Alzheimer's Disease and Related Disorders.
Wiley, Chichester, 93-98.
- Hardy, John A. - Higgins, Gerald A. (1992): Alzheimer's Disease: the Amyloid
Cascade Hypothesis. Science. 256, 184-185.
- Lambert, Mary P. - Finch, C. E. - Krafft, G. A. - Klein, W. L. (1998):
Diffusible, Nonfibrillar Ligand Derived from Abeta 1-42 Are Potent Central Nervous
System Neurotoxins. Proceedings of the National Academy of Sciences of the USA.
95, 6448-6453.
- Laskay Gábor - Zarándi M. - Varga J. - Jost K. - Fónagy A. - Torday C. -
Latzkovits L. - Penke B. (1997): A Putative Tetrapeptide Antagonist Prevents
beta-amyloid Induced Long-term Elevation of [Ca2+]i. Biochemical and Biophysical
Research Communications. 235, 479-481.
- Mager, Peter P. - Penke B. - Walter, R. - Harkany T. - Härtig, W. (2002):
Pathological Peptide Folding in Alzheimer's Disease and Other Conformational
Disorders. Current Medicinal Chemistry. 9, 1763-1780.
- Selkoe, Dennis J. (1991): The Molecular Pathology of Alzheimer's Disease.
Neuron. 6, 487-498.
- Soto, Claudio - Kindy, M. S. - Baumann, M. - Frangione, B. (1996):
Inhibition of Alzheimer's Amyloidosis by Peptides that Prevent beta-sheet
Conformation. Biochemical and Biophysical Research Communications. 226, 672-680.
- Tanzi, Rudolph E. - McClatchey, A. I. - Lamperti, E. D. - Villa-Komaroff, L.
- Gusella, J. F. - Neve, R. L. (1988): Protease Inhibitor Domain Encoded by an
Amyloid Protein Precursor Mrna Associated With Alzheimer's Disease. Nature.
331, 528-530.
- Tariska Péter (2000): Alzheimer-kór. Golden Book, Budapest
- Tjernberg, Lars O. - Naslund, J. - Lindquist, F. - Johansson, J. - Karlstrom,
A. L. Thyberg, J. - Terenius, L. - Nordstedt, C. (1996): Arrest of beta-amyloid
Fibril Formation by a Pentapeptide Ligand. The Journal of Biological Chemistry.
271, 8545-8548.
- Vassar, Robert (1999): Beta-secretase Cleavage of Alzheimer's Amyloid Precursor
Protein by the Transmembrane Aspartic Protease BACE. Science. 285, 735-741.
Kulcsszavak: Alzheimer-kór, Parkinson-kór, Huntington-kór, Amiotrof lateral
sclerosis, Lewy-testes demencia, Prion-betegség, fehérje aggregáció,
béta-szerkezet, gyógyszerkutatás
<-- Vissza a 2003/5 szám tartalomjegyzékére
<-- Vissza a Magyar Tudomány honlapra
[Információk] [Tartalom] [Akaprint Kft.]