1. ábra * A biológiai és bioinformatikai adatbázisok tudományos nyelve elemek és komplex relációk hierarchikus rendszerére épül (A), szemben a tradicionális természetleírás fogalmi rendszerével (B).
Az elmúlt évtized leglátványosabb fejlődése kétségtelenül a számítógéptudományok és a biotechnológia területén zajlott le. Nem meglepő, hogy megjelentek a biológiai információ kezelésének sajátos számítógépes közelítései is, melyeket ma együttesen bioinformatikának szokás nevezni. Ennek a szakterületnek alig tíz-tizenöt év alatt tekintélyes önálló intézményei, folyóiratai létesültek, az egyetemek többségén tantárgyként vagy önálló szakként oktatják, évente több kongresszust rendeznek a témában, és sorra jelennek meg a bioinformatikai tankönyvek is.1 A bioinformatika fogalma azonban nem írható körül módszertani alapokon, már csak azért sem, mert informatikai eszközöket a tudomány minden ágában használnak. Ez az írás amellett igyekszik érvelni, hogy a bioinformatika elsősorban nem különálló szaktudomány, hanem sokkal inkább egy újszerű ismeretábrázolási módszereket alkalmazó általános szemléletmód, melynek fejlődését itt a molekuláris biológia szemszögéből igyekszünk nyomon követni.
A vizsgált objektum szempontjából a bioinformatika három, többé-kevésbé jól elhatárolható irányzatra oszlik, egyik a DNS és a fehérjék szekvenciaadataival, másik a molekulák háromdimenziós szerkezetével, a harmadik pedig a biológiai kölcsönhatások hálózataival foglalkozik. Kezdetben a szekvenciaanalízis fejlődése volt talán a leglátványosabb. A genomadatok ugyanis szekvenciák (karaktersorozatok) alakjában jelennek meg az adatbázisokban, ezek az eredmények a felhasználók széles köre számára érthetőek. Ráadásul a szekvencia nagyon jól kezelhető adattípus. Ebben a körben nagyon sok olyan feladat is megoldható, amely például gráfok vagy háromdimenziós szerkezetek esetében túlságosan időigényes lenne.
Igen eltérőek a háromdimenziós szerkezeti adatokkal foglalkozó számítások motivációi. A szerkezeti bioinformatika történetileg nem a molekuláris biológiából, hanem a makromolekuláris szerkezetkutatásokból (structural biology) fejlődött ki. Ez utóbbi komplex fizikai vizsgálati módszereket (például röntgendiffrakciót és magmágneses rezonancia-spektroszkópiát) és modellezési technikákat (például molekuláris mechanikát, molekuláris dinamikai modelleket) alkalmaz, melyek általában távol esnek a biológusok, orvosok érdeklődésétől.
A kölcsönhatási hálózatok vizsgálata a legújabb kutatási irányzat. Megjelentek ugyanis olyan kísérleti technikák (például microarray és kettőshibrid módszerek stb.), amelyek tömegesen állították elő a gének, fehérjék kölcsönhatási adatait, és ezek ábrázolására a hálózatszerű, gráfelméleti leírások a legkézenfekvőbbek. A három modelltípusban mindössze annyi közös, hogy a biológiai struktúrákat mindegyikük elemek és köztük lévő relációk segítségével írja le (Pongor, 1988), mely utóbbiak egy bonyolult és fejlődő fogalmi rendszert képeznek (1. ábra).
E rövid bevezetés sejteti, hogy a technikai újítások mellett a bioinformatikában az ismeretek ábrázolásának új útjairól van szó. Mindennek látható jele, hogy az új biológia eredményei már egyre inkább elektronikus adatbázisok formájában jutnak el a felhasználókhoz (1. táblázat). Míg azonban a nyomtatott információt saját magunk értelmezzük, az adatbázisokat már közvetve, programok segítségével olvassuk, és a programok saját maguk is sok - a felhasználó számára sokszor korántsem transzparens - információt tartalmaznak. A programokba épített interpretációs módszerek eleinte magában a programkódban voltak elrejtve, újabban azonban már a biológiában is megjelentek az egyes területek szabályait formálisan leíró biológiai ontológiák (objektumokra és viszonyaikra vonatkozó enciklopédikus összegzés [a szerk.]) (2. táblázat). Az alábbiakban a molekuláris ismeretek ábrázolásának nyelvi, térbeli és hálózati modelljeit, ill. a számítástechnikai eszközök fejlődését tekintjük át.
Szekvenciák és nyelvek
Nyelvi leírásoknak tágabb értelemben azon leírásokat nevezzük, amelyek szemantikus, fogalmi definíciók segítségével definiálják az elemeket és a közöttük lévő relációkat. A molekuláris biológia maga olyan korszakban született, mikor a tudományban a nyelvi megközelítések uralkodtak. Ilyenek voltak elsősorban a nyelvelmélet (lásd Ludwig Wittgenstein, Ferdinand de Saussure, Noam Chomsky műveit) és az információelmélet eredményei, valamint a kor katonai kutatásain alapuló kriptológiai és alakfelismerési irányzatok. Ezekre alapozva kialakult egy tág, nyelvi, kommunikációs és számítástudományi eszközöket egyesítő gondolkodásmód, melynek alapján kézenfekvőnek tűnt, hogy maguk a biológiai szekvenciák is egyfajta kódot jelentenek, azaz egy ismeretlen nyelven leírt információ hordozói. Ez a metaforikus értelmezés olyan kifejezésekben köszön vissza, mint a "genetikai kód" vagy "az élet könyve".
A biológiai szekvenciák elemzése először valóban a matematikai nyelvészet karaktersorozat-elemzési metodikáit használta, ezek közül sokan, például a sorozatok komplexitási vizsgálatai, ma is a standard módszerek közé tartoznak. Mikor a 90-es években megindultak a genomszekvenálási programok, a karaktersorozatok hasonlóságait vizsgáló heurisztikus módszerek - például a jól ismert BLAST program (Altschul et al., 1990) - lettek a leggyakrabban alkalmazott algoritmusok, és nemcsak a bioinformatika, hanem az egész tudományos számítógép-felhasználás területén.
A szekvenciák adatbázisainak története a 60-as években kezdődött, mikor Margaret Dayhoff és munkatársai a Georgetown Egyetemen rendszerezni kezdték és közzétették az irodalomból gyűjtött fehérjeszekvenciákat. A példaértékű vállalkozás azonban hamar kinőtte a nyomtatás lehetőségeit, át kellett térni a kizárólagosan számítógépes tárolásra. Közben megjelentek a DNS-szekvenálás produktív technikái is, melyek az addig egyeduralkodó fehérjeszekvenálási módszereket lassan ki is szorították a tömeges adatgyűjtésből. Az adatok ma már szinte kizárólag DNS-szekvenálásból származnak, gyűjtésük nemzetközi kollaboráció keretében folyik. Erre a célra az Egyesült Államok, az Európai Unió és Japán szakintézményeket hozott létre. A 90-es években jelentkezett két új fejlődési irány. Egyrészt megjelentek az első specializált adatbázisok - ilyen volt például az első fehérje doménszekvencia gyűjtemény (Pongor et al., 1993) - amelyek a szekvenciák egyes előre csoportosított szakaszaira koncentráltak. Ezek száma ma már meghaladja a százat. Másrészt az adatbázisok annotációja is egyre rendszeresebbé vált. Eleinte ugyanis a szekvenciák mellett csak egy azonosító kód állt, később megjelentek a bibliográfiai, funkciós és szerkezeti információk, illetve a bibliográfiai adatbázisokra történő hivatkozások is. Mindezek az információk természetes nyelven íródtak. Hamar felmerült tehát egy uniform, számítógéppel olvasható tudományos nyelv iránti igény. A természetes nyelvet használó megközelítések eredményei egyrészt a biológiai ontológiák, másrészt a bibliográfiai adatbányászat módszerei, melytől a nagy számban produkált molekuláris adatok értelmezésének automatizálását várják.
Térbeli modellek
A térbeli látásmód csírái már az atomokra vonatkozó legkorábbi elképzelésekben tetten érhetők, a modern molekuláris tudományokban pedig talán Jacobus Henricus van t'Hoff volt az első, aki 1874-ben felismerte, hogy egyes kémiai tulajdonságok nem magyarázhatók az atomok háromdimenziós elrendeződése nélkül (Palló, 2001). Ahogy a röntgendiffrakció módszerei fokozatosan lehetővé tették a szerves molekulák vizsgálatát, Olga Kennard és John Desmond Bernal elkezdték a molekuláris szerkezetek rendszerezését, ebből a gyűjteményből fejlődött ki a későbbi Cambridge Structural Database adatbázis, mely nagyban hozzájárult ahhoz, hogy a kémiában konszolidálódtak a molekuláris geometria és a sztereokémia fogalmai. A makromolekulák szerkezeti adatbázisainak közös őse az 1971-ben alapított Protein Data Bank (Bernstein et al., 1977). A 90-es évek során a háromdimenziós adatokra vonatkozó adatbázisok között is megjelentek a másodlagos adatbázisok, melyekből szintén több tucatot számlálhatunk össze (Carugo - Pongor, 2002). A nukleinsavak szerkezeti adatbázisai a közelmúltig a fehérjékétől eltérő utakon fejlődtek. A nukleinsavak háromdimenziós szerkezetmeghatározása ugyanis gyakorlatilag sokkal nehezebb és kevésbé produktív, mint a fehérjéké, és a meglévő adatok többsége is rövidebb DNS-szakaszokra vonatkozik. Mind fehérjéknél, mind nukleinsavaknál alkalmazunk alacsonyfelbontású modelleket is, a DNS makroszkopikus alakváltozásait szokás például végtelen szál, pontosabban elasztikus rúdmodell segítségével ábrázolni (Munteanu et al., 1998).
Ahogy a szekvenciák számára a nyelv, a háromdimenziós modellek metaforái számára a tárgyakra vonatkozó ismeretek jelentik a fogalmi hátteret. A tárgyak kezelésének képessége pedig, ha lehet, még ősibb tulajdonság, mint a nyelvi készség. A számítógépes grafika a makromolekulák szerkezetét a tárgyi világból kölcsönzött objektumok, például hengerek, gömbök, szalagok, felületek segítségével jeleníti meg, ezek révén az áttekinthetetlen atomi részletek helyett kirajzolódnak a makromolekulák formái, térbeli mintázatai, szimmetriái. A fehérjék konformációjának analízise, a fold-típusok felismerése például lehetetlen lett volna számítógépes vizualizálási módszerek nélkül, de a tárgyi metafora megjelenik a szerkezeti számításokban is, hiszen a molekulaszerkezetek optimalizálásakor a szerkezetet például gyakran egyszerű, klasszikus mechanikai tárgyakként modellezzük. Végül, valahol a háromdimenziós leírási módszerek határán helyezkednek el az egy- vagy többdimenziós diagrammatikus ábrázolások, melyekben az adott molekulák valamilyen jellemzőjének eloszlását ábrázoljuk (Pongor, 1987).
Hálózati modellek
Az előbbi két modelltípusnál kikötöttük, hogy az elemek és kapcsolataik definíciói a tudományos nyelv szemantikai, illetve a molekuláris geometria tárgyi leírásait kövessék. Ha ezektől a megkötésektől eltekintünk, általános topológiai modellekhez, vagyis hálózatokhoz jutunk, amelyekkel tetszőleges elemek és viszonylatok ábrázolhatók, mint például a metabolikus útvonalak, a neuronok hálózatai, a táplálkozási láncok, gének regulációs hálózatai. Ennek az egyszerű ábrázolásmódnak első példája a kémiában alkalmazott szerkezeti képlet, matematikai formája pedig a gráf - például egyszerű, irányított és súlyozott gráf. A molekulaszerkezetek egyébként már a XIX. században is inspirálták a gráfelmélet fejlődését. Fontos lépés volt az Erdős Pál és Rényi Alfréd nevéhez fűződő véletlen-gráf modell (Erdős - Rényi, 1959), amelyet később a sejten belüli hálózatok állapotainak modellezésére alkalmaztak (Kauffman, 1992). A hálózati modellek napjainkbeli népszerűsége viszont annak a felismerésnek köszönhető, hogy egyes biológiai és technikai hálózatok (például Internet, légiutak stb.) tényleges topológiája jellemzően eltér a véletlen gráfoktól, és ez összefüggésbe hozható a hálózatok stabilitásával (Barabási - Albert, 1999). A hálózati modellek különösen hasznosnak bizonyultak az új technikák, például a genomszintű expressziós vizsgálatok eredményeinek ábrázolásában, itt ugyanis a nagy adattömegből a kölcsönható elemek éppen részgráfok segítségével azonosíthatók.
Számítógépes módszerek
A bioinformatika olyan korszakban keletkezett, mikor a számítástechnika bevonult a biológiai kutatások mindennapos gyakorlatába. Adatbázis-architektúrák, programozási módszerek, felhasználói felületek és más szoftverkomponensek tekintetében a bioinformatika lényegében nem különbözik az informatika egyéb területeitől. Önálló színt hoztak a nyolcvanas évek közepén megjelenő bioinformatikai programcsomagok, amelyek egységes, moduláris rendszerben tudták kezelni a szekvenciákkal kapcsolatos módszerek majdnem teljes körét. Ezek a programok eleinte az egyetemek központi számítógépei révén terjedtek el, ma már személyi számítógépen is hozzáférhetők. Közben kialakultak a biológiai adatok rögzítésének standardizált formátumai is, így a mai felhasználó az adatbázisokból letöltött adatokat már könnyen tudja analizálni. A kereskedelmi programcsomagok nehezen tudnak lépést tartani az újabb és újabb módszerekkel, ezért megjelentek a programcsomagok, programkönyvtárak nyílt forrású (open source) változatai, amelyek - a Linux rendszerhez hasonlóan - a fejlesztők önkéntes kooperációján alapulnak. Az egyetemi, kutatóintézeti csoportok pedig legtöbbször közvetlenül, internetes szolgáltatás formájában teszik közzé új módszereiket. Ilyenkor a számítás részletei ugyan gyakran rejtve maradnak, de a felhasználók biztosak lehetnek abban, hogy a program legújabb változatához férnek hozzá. A módszerfejlesztők is kedvelik a webszervereket, mert így munkájuk azonnal ismertté válik,2 míg a programcsomagokba illesztett módszerek szerzőire kevesebbet hivatkoznak.
A leglátványosabb fejlődést azonban kétségkívül a World Wide Web technika okozta és okozza. Ennek segítségével először is az adatbázisok közötti keresztreferenciák "navigálhatóvá" váltak, így ma a felhasználó pillanatok alatt tud váltani ismerettípusok és adatbázisok között. A biológiai adatbázisok hálózata tényleges hasonlóságot mutat az emlékezet asszociációs hálózataival, azzal a különbséggel, hogy itt a kapcsolatok - legalábbis egyelőre - statikusak és előre kiszámítottak. Egy adatbázis elemre nézve - ami lehet például szekvencia, térszerkezet, szöveges összefoglaló - időről időre meghatározzuk a hozzá valamilyen szempontból hasonló elemek körét, és ezek között WWW (World Wide Web) kapcsolatot létesítünk. Így ún. "szomszédságok" keletkeznek, és a felhasználó ezeken keresztül navigálva egy DNS-szekvenciától eljuthat például egy térszerkezethez vagy a biológiai funkció szöveges összefoglalójához, a kódolt fehérje metabolikus vagy evolúciós környezetének térképéhez (2. ábra). Az ilyen integrált rendszerekben általában ingyenesen hozzáférhetőek az analízis számításos módszerei (WWW szerverek), de egyre gyakrabban találunk elektronikus oktatási anyagokat és tankönyveket is. Kialakulóban van tehát egy bioinformatikai infrastruktúra, ahol a felhasználók mélyreható elméleti vagy számítástechnikai ismeretek nélkül is el tudnak végezni majdnem minden rutinfeladatot. Az igényesebb publikációkhoz azonban továbbra is szükség van a helyben telepített programokra is. Ezzel párhuzamosan a bioinformatika felhasználóinak köre is www-orientált rutinfelhasználókra, és a programozásban jártas szakfelhasználók körére oszlik; utóbbi felhasználókat a biomedikai kutatás-fejlesztés legtöbb területén különösen szívesen alkalmazzák.
Az ismeretábrázolás új szintje
A bioinformatika - bár képviselői gyakran panaszkodnak az adatok özönvízszerű áradására - nem mozgat több adatot, mint a tudomány és a technika többi ágazata. A különbséget sokkal inkább az adatok sokféleségében és az ismeretek komplexitásában kell keresnünk. Az új technológiák és számítási eszközök révén ugyanis sok, eddig hozzáférhetetlen probléma lett kísérletileg vizsgálható, s így az élettudományokban a komplex fogalmak és megközelítések egész sora alakult ki (elegendő például a funkcionális genomikára, a szerkezeti biológiára gondolnunk). Nem túlzás, hogy a biológiai, molekuláris és bibliográfiai adatbázisok hálózata talán a legösszetettebb tudásábrázolási rendszer a tudomány eddigi történetében.
Mi okozhatta a komplex ismeretek új hullámát? Biológiáról lévén szó, talán helyénvaló egy evolúciós hasonlat. Szathmáry Eörs és John Maynard Smith az egyre komplexebb életformák megjelenését a genetikai információ tárolásának és kommunikációjának megváltozásával, mint például a kromoszómák megjelenésével, az eukarióták és a többsejtű organizmusok kialakulásával magyarázták (Szathmáry - Smith, 1995; Smith - Szathmáry, 1995). Az ismeretábrázolás komplexitási ugrásait ennek megfelelően az ismeretek tárolásának és kommunikációjának megváltozásában kell keresnünk. A tradicionális társadalmakban szóban kommunikálják az ismereteket, melyeket aztán egy egész emberi közösség tárol és örökít. Összetett ismeretek rögzítésének és kommunikációjának lehetőségét az írásbeliség teremtette meg, de ez tömegessé csak a könyvnyomtatás - azaz egy új kommunikációs technológia - révén válhatott. Ekkor alakultak aki a tudás tárolásának szervezett és szisztematikusan kerestethető hordozói, az enciklopédiák (Nyíri, 2003), amelyek sok tekintetben a mai elektronikus adatbázisok előfutárainak tekinthetők. A bioinformatika kialakulásakor mind az elektronikus adattárolás, mind pedig az adatokkal való kommunikáció megváltozott, s ez utóbbiban véleményünk szerint az adatok széles körének hálózatba történő összekötése volt az a lényeges lépés, amelyben az élettudományok talán némileg megelőzték a többi alkalmazási területet. Összefoglalásként elmondhatjuk, hogy az új kommunikációs módszerek révén a biológiában egy új, komplexebb szemléletmód van kialakulóban, melynek alapjai a nyelvi, vizuális és hálózati modellek.
Ezúton szeretném megköszönni Csányi Vilmos, Falus András, Izsák János, Nyíri Kristóf, Pléh Csaba és Szathmáry Eörs segítségét, tanácsait és értékes gondolatait, illetve az Oktatási Minisztérium támogatását (OMFB -01887/2002, OTKA M45378).
Kulcsszavak: bioinformatika, molekuláris biológia, adatbázis
1 Magyar nyelven olvasható Malcolm A. Campbell és Laurie J. Heyer Genomika, proteomika, bioinformatika című orvosbiológiai szemléletű bevezető tankönyve (Medicina, Budapest, 2004). Magyar nyelvű oktatási anyagok találhatók például a www.brc.hu/ps/bioinformatika-oktatas címen.
2 Pl. az ISI felmérése ( www.in-cites.com/countries/hungary.html) szerint az elmúlt 10 év legtöbbet idézett magyar szerzőségű cikke is egy webszerveren közzétett számítási módszer (Tusnády - Simon, 1998).
Rendszer Elem Reláció Molekulák, pl. Atomok Kémiai kötés fehérjeszerkezet Fehérje- Aminosavak Szekvenciális szekvencia szomszédság Egyszerűsített Másodlagos Szekvenciális fehérjeszerkezet szerk. elemek szomszédság, 3-D Fold-típusok Ca-atomok Szekvenciális szomszédság, 3-D Komplexek Fehérjék, DNS Molekula- kölcsönhatások Metabolikus Enzimek Kémiai reakciók útvonalak (szubsztrát/termék) Génhálózatok Gének Ko-reguláció
1. ábra * A biológiai és bioinformatikai adatbázisok tudományos nyelve elemek
és komplex relációk hierarchikus rendszerére épül (A), szemben a tradicionális
természetleírás fogalmi rendszerével (B).
Nukleotid-szekvencia adatbázisok Nemzetközi nukleotid-adatbázis kollaboráció (USA-EU-Japán) Kódoló és nem kódoló DNS Génszerkezet, intronok, exonok Transzkripciós faktorok és kötőhelyeik RNS szekvenciák Fehérjeszekvencia-adatbázisok Általános szekvencia adatbázisok Fehérjetulajdonságok Fehérjelokalizáció és targeting Fehérjeszekvencia-motívumok és aktív helyek Fehérjedomének, fehérjék osztályozása Egyes fehérjecsaládok adatbázisai Szerkezeti adatbázisok Kis molekulák Szénhidrátok Nukleinsav-szerkezetek Fehérjeszerkezetek Genomadatbázisok (nem-gerincesek) Genomannotációs kifejezések, ontológiák és nevezéktan Taxonómia és azonosítás Általános genomadatbázisok Vírusgenomok Prokaióta-genomok Egysejtű eukarióta genomok Gombagenomok Egyéb nem-gerinces genomok Metabolikus és jeltovábbbítási (signaling) útvonalak Enzimek és enzim-nevezéktan Molekuláris kölcsönhatások és jeltovábbítási útvonalak Emberi és egyéb gerinces genomok Modellszervezetek, összehasonlító genomika Humán genom adatbázisok, géntérképek és vizualizáló eszközök Humán ORF-ok Humán gének és betegségek Microarray és egyéb génexpressziós adatbázisok Proteomikai adatbázisok és analizáló rendszerek Egyéb molekuláris biológiai adatbázisok Biológiai makromolekulák képei Bioremediációs adatbázisok Gyógyszerek és gyógyszertervezés Jelzőmolekulák és primerek Organellum adatbázisok Növényi adatbázisok Általános növényi adatbázisok Arabidopsis thaliana Rizs Más növények Immunológiai adatbázisok1. táblázat * A bioinformatikai adatbázisok főbb típusai (A Nucleic Acids Research 2005. évi adatbázisszáma alapján, http://www3.oup.co.uk/nar/database/cat/12/.
Genew - human gén-nevezéktani adatbázis, http://www.gene.ucl.ac.uk/cgi-bin/nomenclature/searchgenes.pl GO - Gene Ontology, gén-ontológia, http://www.geneontology.org/ GOA - Gene Ontology Annotation, gén-ontológia, http://www.ebi.ac.uk/GOA IUBMB Enzim-nevezéktani adatbázis, http://www.chem.qmul.ac.uk/iubmb/ IUPAC Szerves- és biokémiai nevezéktan, http://www.chem.qmul.ac.uk/iupac/ IUPHAR-RD Gyógyszerek és receptorok farmakológiai adatbázisa, http://www.iuphar-db.org/iuphar-rd/ PANTHER Géntermékek nevezéktana, http://panther.celera.com/ STAR/mmCIF: an ontology for macromolecular structure, http://ndbserver.rutgers.edu/mmcif UMLS - Unified Medical Language System (Egységes orvostudományi nyelvrendszer, tezaurusz, lexikon és szemantikus hálózatok formájában) - http://umlsks.nlm.nih.gov
2. tábl. * Az adatbázis-annotációk nyelvezetének és nómenklatúrájának adatbázisai, ontológiák
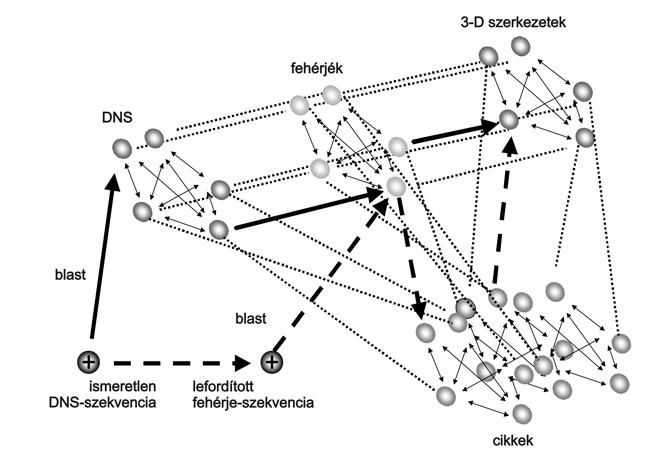
2. ábra * Keresés integrált molekuláris adatbázisokon. Az egyes adatbázisok
(DNS- és fehérje-szekvenciák, 3-D szerkezetek, szakcikkek) WWW
kereszthivatkozásokkal vannak ellátva (pontozott vonal). Az egyes adatbázisokon
belül további kapcsolatok (vékony nyilak) kötik össze a hasonló tételek
"szomszédságait". Így ha egy hasonlóságkeresési módszer (vastag nyíl) egy
nem-annotált (ismeretlen funkciójú) DNS-szekvenciára mutat, akkor ugyanazon
szekvencia szomszédságának egy (nagy valószínűséggel funkcionálisan rokon)
másik, annotált (már jobban ismert) tagja segítségével az adatgyűjtés során
továbbléphetünk a fehérjék, esetleg a 3-D szerkezetek irányában. Ezekben a
példákban a keresést itt a BLAST programmal kezdtük, de más pontokon is
kezdhetünk.
Irodalom
Altschul, Stephen F. - Gish, W. - Miller, W. - Myers, E. W. - Lipman, D. J. (1990): Basic Local Alignment Search Tool. Journal of Molecular Biology. 215, 403-410.
Barabási Albert László - Albert Réka (1999): Emergence of Scaling in Random Networks. Science. 286, 509-512.
Bernstein, Frances C. - Koetzle, T. F. - Williams, G. J. - Meyer, E. F. Jr. - Brice, M. D. - Rodgers, J. R. - Kennard, O. - Shimanouchi, T. - Tasumi, M. (1977): The Protein Data Bank: A Computer-Based Archival File for Macromolecular Structures. Journal of Molecular Biology. 112, 535-542.
Carugo, Oliviero - Pongor Sándor (2002): The Evolution of Structural Databases. Trends Biotechnology, 20, 498-501.
Erdős Pál - Rényi Alfréd (1959): On Random Graphs. Publicationes Mathematicae Debrecen. 6, 290-297.
Kauffman, Stuart A. (1992): The Origins of Order: Self-Organization and Selection in Evolution. Oxford University Press, Oxford, UK
Maynard Smith, John - Szathmáry Eörs (1995): The Major Transitions in Evolution, W.H. Freeman, Oxford, New York, Heidelberg
Munteanu, Mircea G. - Vlahovicek, K. - Parthasarthy, S. - Simon I. - Pongor S. (1998). Rod Models of DNA: Sequence-Dependent Anisotropic Elastic Modelling of Local Bending Phenomena. Trends Biochemical Sciences. 23, 341-347.
Nyíri Kristóf (2003): Enciklopédikus tudás a 21. században. Mindentudás Egyeteme, 2003. dec. 21.
Palló Gábor (2001): Vizualitás és a kémia nyelve. Világosság. 7-9, 58-66.
Pongor Sándor (1987): The Use of Structural Profiles and Parametric Sequence Comparison in the Rational Design of Polypeptides. Methods in Enzymology. 154, 450-73.
Pongor Sándor (1988): Novel Databases for Molecular Biology. Nature. 332, 24.
Pongor Sándor - Skerl, V. - Cserző M. - Hátsági Z.- Simon G. - Bevilacqua, V. (1993): The SBASE Domain Library: A Collection of Annotated Protein Segments. Protein Engineering. 6, 391-395.
Szathmáry Eörs - Maynard Smith, John (1995): The Major Evolutionary Transitions. Nature. 374, 227-232.
Tusnády Gábor E. - Simon István (1998): Principles Governing Amino Acid Composition of Integral Membrane Proteins: Application to Topology Prediction. Journal of Molecular Biology. 283, 489-506.