Számítógépes modellek
a molekuláris orvostudományban
és bioinformatikában
– alkalmazás és a távlatok
Vladimir Brusic
Australian Centre for Plant Functional Genomics,
School of Land and Food Sciences, and Institute for Molecular Bioscience,
University of Queensland Australia
v.brusic @ uq.edu.au
Bevezetés
A molekuláris orvostudomány a diagnózisra, gyógykezelésre, sérülések és betegségek megelőzésére összpontosít, hogy a kóros eltéréseket molekuláris szinten is megtalálja. A molekuláris orvostudományi kutatások fontos céljai a DNS vagy fehérjesorrendről kapott információból eredő diagnosztikai tesztek, gyógyászati eljárások, illetve profilaktikumok. A kutatás ezen a területen főleg experimentális jellegű, de ezek gyakran megfizethetetlenül drágák vagy nem is lehetségesek. A számítógépes modellezés a kísérleti kutatást támogató, kiegészítő technológiaként került előtérbe. Ez különösen hasznos olyan esetekben, amikor a tanulmányozott probléma természete több ezer vagy akár több millió egyedi kísérletet kívánna. A molekuláris kölcsönhatások pontos modelljeit alkalmazó előzetes szűrés segíthet felismerni néhány olyan kulcsfontosságú kísérletet, mely elegendő a tanulmány befejezéséhez.
Nagyteljesítményű technológiák, mint például a genomika vagy proteomika, a gén és fehérje-expressziókról hatalmas mennyiségű adatot állítanak elő (Auffray et al., 2003). A molekuláris folyamatok kombinatorikai jellege szükségessé teszi a kísérletekkel együtt történő számítógépes modellezés integrálását olyan molekuláris orvosbiológiai tanulmányokba, mint például a transzkripciós szabályozás (Beer és Tavazoie, 2004), vagy az antigénfeldolgozás és prezentáció (Flower, 2003). A számítógépes modellezés hasznos a kísérletek megválasztása és tervezése során, valamint az összegyűlt adatokból levont következtetések értelmezésében és megértésében is (Brusic – Zeleznikow, 1999).
Ebben a cikkben a számítógépes modellek kifejlesztésének és felhasználásának követelményeit tárgyaljuk, illetve bemutatjuk azok fejlődését egy adott terület adatainak összegyűjtésével. Ezt olyan immun epitópok (antigéndeterminánsok) azonosításának példáit használva szemléltetjük, melyek az oltóanyagfejlesztés célterületéhez tartoznak.
A számítógépes modellek
felépítésének követelményei
Maximális kihasználtsághoz a számítógépes modellek teljesítményét még használat előtt ki kell alakítani és meg kell becsülni; ezt a becslést ugyanolyan szigorúsággal kell elvégezni, mint a standard laboratóriumi folyamatok esetében. Ez a tesztelésben a megfelelő gondosságot, a számítógépes modellek validálását, a szimulált kísérletek megtervezését és az eredmények értelmezését jelenti. A számítógépes modellek tervezéséhez és kifejlesztéséhez a legjobb modellező és komputeres alkalmazásokat kell használni. Használat előtt meg kell állapítani a számítógépes modellek relevanciáját, pontosságát, általánosító képességét, precizitását és teljesítőképességét. A releváns és pontos számítógépes modellek alkalmazhatók a laboratóriumi kísérletek kiegészítéseként, és alkalmazhatók számítógépes vizsgálatok céljából is (Brusic – Zeleznikow, 1999). A modell-relevancia kifejezés a feltételezések helyességére utal – melyeknek összhangban kell lenniük a tanulmányozott problémával kapcsolatos legmodernebb tudományos és technikai ismeretekkel. Például a szignál peptideket becslő modellek nem használhatók a fehérjék transzmembrán területeinek beazonosításához. A predikciós modellek számos pontossági mérési mutatója ismert (Bajic, 2000). A gyakran használt mutatókat az 1. táblázat mutatja.
Az általánosító képesség a modell azon képességét jelzi, hogy mennyire pontosan tudja megjósolni az új eseteket, vagyis a modell gyakorlati használhatóságát mutatja. A gyenge általánosító képesség a következőkből adódhat: a) nem megfelelő adathalmaz a betanítás idején, b) alkalmatlan tanuló algoritmusok kiválasztása és használata, c) a modell komplexitása nem illik a modellált jelenséghez vagy a begyakorláshoz használt adathoz.
Amikor a modellépítéshez használt adatok túlspecializáltak, pl. a releváns adatoknak csak egy részhalmazát jelképezik, a modell nem biztos, hogy megfelelő a tervezett használatra. Például a globuláris fehérjékből származó fehérje szerkezeti modellek alig, vagy nem megfelelően alkalmasak, ha a fehérjék transzmembrán területei szerkezetének predikciójára használják őket. Gyenge általánosítás adódhat a modell túlértékeléséből is – olyan helyzetben, amikor a modellt a tanuló adatbázis sajátosságainak megtanulására kényszerítik a modellált rendszer általános szabályainak megtanulása helyett. Végül, a túlságosan bonyolult modellek hajlamosak memorizálni a gyakorló példákat, az általános szabályokat pedig gyakran nem. A túl egyszerű modellnek viszont korlátozott képessége lesz a tanulásra, mely a predikciók nagyobb pontatlanságát eredményezi.
A számítógépes modellek alkalmazásának rendszere
A modellezést magába foglaló orvosbiológiai tanulmányok különböző szinteken végezhetők – molekuláris, sejt-, szervi, szöveti, szervezeti vagy populációs szinteken (Motta – Brusic, 2004). Itt nem tárgyaljuk a szubmolekuláris és ökoszisztémabeli szinteket, bár néhány esetben ezeknek is van relevanciájuk az egészséges vagy beteg állapotok magyarázatában. A molekuláris szintű tanulmányokban erőteljes fejlődés tapasztalható a DNS, RNS, fehérje- és szénhidrátszerkezetek háromdimenziós modelljeinek alkalmazásától kezdve a metabolikus vagy szabályozó folyamatokban részt vevő molekuláris hálózatának modellezéséig. Ezeket a modelleket a potenciális gyógyszertargetek molekuláris kiválasztásában és azonosításában alkalmazzák – ilyen például sok más mellett a szignál peptidek predikciója (Nielsen et al., 1999), a transzkripciós kötődési helyek azonosítása (Tompa et al., 2005), az antigének feldolgozása (Petrovsky – Brusic, 2004). Molekuláris szintű folyamatok vesznek részt a sejtes folyamatok beindításában és szabályozásában, melyeket a sejtszintű modellek képviselnek.
A sejtszintű modellek sejtállapotokat írnak le, melyek kulcsfontosságúak például az allergiához, autoimmunitáshoz, tumorhoz és fertőző betegségekhez vezető folyamatok megértésében. Az ilyen modellek döntő fontosságúak a terápiás megközelítés megtervezéséhez és molekuláris szintű modellek kiterjesztésében – így például a sejtszignálok in silico modelljei (Recanatini et al., 2005), az ún. „E-sejt projekt” a teljes sejt modellezésére (Takahashi et al., 2003), illetve a mikroorganizmusok modelljei (Ishii et al., 2004). A szervi és szöveti modellek a modellezést az orvostudomány klasszikus felosztásához és a szervek és szövetek fiziológiai, elektrofiziológiai, mechanikus, biokémiai és funkcionális leírásához közelítik. E modellek a diagnosztikát, a gyógyászati alkalmazásokat támogatják, és az orvosok széleskörűen alkalmazzák őket. Segítik a folyamatos ellenőrzést (Scher et al., 2005), a manuális orvosi beavatkozásokat (Suzuki et al., 2005), illetve a terápiás kezelésekben való döntéshozást. A példák között szerepelnek az agytérképek (Nowinski et al., 2005), szív- és tüdőmodellek (Lador et al., 2005), a thymusfejlődés modellje (Wang et al., 2004) és sok más. A szervi és szöveti modellek kulcsfontosságúak a modern diagnosztikai szemléletben (Reeves és Kressler, 2004; Muller et al., 2004), illetve a korszerű terápiák meghatározásában (Fraser és Rappuoli, 2005). Az élőlények roppant mértékű bonyolultsága, illetve a teljes szervezet egyedisége miatt a magasabb rendű élőlények matematikai és számítógépes modelljeinek napjainkban korlátozott a felhasználhatósága. Ilyen kezdeményezések többek között a Humán Genom Project (Roberts et al., 2001), a „Látható ember” projekt (Banvard, 2002), vagy a nemrég kezdeményezett ImmunoGrid/Immunhálózat – mely az emberi immunrendszert modellálja a hálózati („grid”) számítás révén (Brusic et al., közlés alatt). Egyes projectek integrált megközelítésre összpontosítanak, például a Physiome project (Hunter et al., 2005), mely integrálni kívánja a molekuláktól elkezdve minden szervrendszer strukturális és funkcionális adatait, mintázatait, számítógépes eszközeit és interneten hozzáférhető adatbázisait. Az adatok értelmezésében a populációra épülő tanulmányok hagyományosan statisztikai alkalmazásokra építenek, illetve a modellek a tendenciák/irányvonalak megjóslásában és a populáció alapú adatok megértésében fontosak. A populáció alapú modellek alkalmazása nagyon sokféle, és normális esetben túlmutat e cikk keretein, kivéve talán a molekuláris vonatkozásúakat. A példák egy része a szubpopulációk egészségének egyenlőtlen eloszlását modellálja elemzési célokból (Reifsnider et al., 2005), a betegségek terjedését, felügyeletét, a megfelelő közegészségügyi intézkedéseket tartalmazza (Matthews – Woolhouse, 2005), megbecsüli a gyerekeknek adható optimális gyógyszeradagolást (Johnson, 2005), illetve a gyakori betegségek komplex jellegzetességeit tanulmányozza (Hirschhorn, 2005).
Az ebben a cikkben felsorolt modellek nem foglalják magukba az összes modellt. Inkább azért használtuk őket, hogy szemléltessük a matematikai és számítógépes modellek bioinformatikában és orvostudományban való használatának kiterjedtségét és mélységeit. A molekuláris folyamatok modellezése a modellezés minden szintjét felöleli; a magas színvonalú orvosi diagnosztika, beavatkozás vagy megelőzés egyre inkább a számítógépes modellek alkalmazásának lesz alárendelve. A számítógépes modellek mindinkább hálózatként kapcsolódnak össze a klinikai megfigyelésekkel és kísérleti megközelítésekkel. Az 1. ábra mutatja a kutatások példáinak szerkezetét, melyeket a számítógépes modellezés segít.
Esettanulmány:
immun epitópok predikciója
Az immunrendszer T-sejtjei rövid peptideket ismernek fel, melyek a fő hisztokompatibilitási komplex molekulákhoz kötődnek (MHC) és a gazdasejtek felszínén helyezkednek el. Ezek a peptidek „felismerési címkék”, melyek a gazdasejt összetételéről tanúskodnak az immunrendszer T-sejtjeinek. A nem homeosztatikus (nem saját) peptidek jelenléte az immunválasz kezdetének előfeltétele. Az intracelluláris fehérjék lebontásából termelt peptidek kötődnek az MHC I. osztályba tartozó molekuláihoz. Az MHC II. osztályú molekulák a peptideket, melyek az extracelluláris eredetű fehérjék lebontásából termelődnek, professzionális antigén-prezentáló sejteken mutatják be. A citotoxikus T-sejtek fő feladata, hogy receptoraikkal (T-sejt receptorok) felismerjék és elpusztítsák a fertőzött (pl. vírus vagy baktérium által), mutáns (pl. tumor) vagy genetikailag idegen (pl. transzplantált) sejteket. Az intracelluláris fehérjék és feldolgozási módjaik felhasználhatósága meghatározza, hogy a.) mely peptidek lesznek hozzáférhetők az MHC I. molekulákkal együttes bemutatásra, b.) mekkora lesz az azt követő citotoxikus válasz. Az MHC II osztály molekuláival együtt bemutatott peptidek főleg az immunválaszok szabályozásában játszanak szerepet; döntő fontosságúak az immunválaszok megkezdésében, fokozásában és gátlásában. Az MHC molekulák peptidkötő helye egy „molekuláris hasadék”/zseb”, mely -hélix-szel kapcsolódni képes -lemezt foglal magába. A peptid a peptidlánc/gerinc és a hasadék közötti hidrogénhíd-hálózaton keresztül kialakuló, illetve a peptid oldalláncai és a hasadék specifikus zsebei közt kialakuló kötésekkel kapcsolódik (Madden et al., 1993). A peptid és a kötőhely közötti kölcsönhatás főként primer és szekunder (elsődleges és másodlagos), a peptiden belüli pozíciók horgonyain keresztül jön létre, így kialakítva az erős kötődést. Egy adott MHC molekula számára csak korlátozott számú aminosav (csoport) tud horgonyként viselkedni egy peptiden belüli konkrét pozícióban. A horgonypozíciók a peptidcsoportokon belüli közös sémák meghatározásának kulcsa, mert ezek kötődnek a specifikus MHC molekulákhoz. Eddig több mint 200, genetikailag eltérő MHC molekula kötő(dési) mintáját közölték (Rammensee et al. 1999). Ezek a minták alapot adnak az MHC-peptid kötések predikciós módszereinek fejlesztéséhez. A 2. táblázatban bemutatunk egy kötődési minta példát. A kötési minták az MHC és peptid között kialakuló kötések alapmodelljeit képviselik, hiszen jelzik egy adott MHC molekulához kötődő peptidekben egy adott pozícióban egy adott aminosav megközelítő preferenciáját. Korábban az ismert MHC-kötődést megjósoló módszerek közül a kötési minták voltak a legpontatlanabbak (Yu et al., 2002).
A mennyiségi mátrix kísérleti adatokból származó, kifinomult kötődési minták ösz-szessége. A finomításhoz az szükséges, hogy egy peptidben megbecsüljük, hogy minden egyes aminosav minden pozícióban meny-nyire vesz részt a kötődésben. A mátrixokat kísérleti adatokból nyerték. Az aminosavak minden pozíciójában kapott adatok összesítése egy kötési eredményt hoz létre, és egy meghatározott küszöbérték felett nagy valószínűséggel kötődő petideket jelent. A HLA-DR4 esetében (Hammer et al., 1994) mind a kötődő, mind a nem kötődő peptidek esetében 70 % feletti a számítógépen helyesnek bizonyult predikció. A mennyiségi mátrixok hatékonyak, könnyen használhatóak, és pontosabbak, mint a kötődési minták. A peptidek és MHC molekulák közti kölcsönhatások nem lineárisak (Yu et al., 2002), míg a mátrixok és a minták csak lineáris kapcsolatokat írnak le. A mesterséges neurális hálózatok (ANN) összetettebbek a minta- vagy mátrix-alapú modelleknél, és több kötődési adatra van szükségük a begyakoroltatáshoz, illetve az adatok előfeldolgozásához. Az elő-feldolgozáshoz peptidkijelölésre/csoportosításra, illetve a formátum az ANN szoftver számára elfogadható átalakítására van szükség. Az egyszerű minták és kötődési mátrixok segíthetik a peptid-előfeldolgozás lépését. Az elsődleges horgonyok információit például az emberi MHC II. osztályú HLA-DR4 molekulához való hamisan pozitív peptidkötődések megbecslésében használták (Brusic et al., 1998). A rejtett Markov-modellek (HMM) valószínűségi (probabilista) vázat használnak, hogy a kutatási hiányosságokat feltérképezzék. Az adatcsoportokból megtudhatják az általánosított probabilista szabályokat. A HMM-eket a HLA-A2 kötő peptidek predikciójában használták (Mamitsuka, 1998). A kifinomult osztályzó modellek egy másik típusát, a segítő vektor gépek (SVM) is alkalmazták az MHC-kötő peptidek tanulmányozásában (Zhao et al., 2003). Ehhez a HMM-eket és ANN-eket kombinálták a véletlenszerűen MHC-kötő peptidek és az antigéneken belüli, immunológiai „forró pontok” predikciójához (Srinivasan et al., 2004). A molekuláris modellezés felöleli az MHC molekulák kristályszerkezetének, illetve a fehérje-peptid kölcsönhatások részletes ismeretét, és arra használják őket, hogy megbecsüljék az MHC molekulákhoz való peptidkötődést (Schafroth – Floudas, 2004). A 3D molekuláris modellek pontosságát még fejleszteni kell, mielőtt nagyarányú új predikciókra használhatnánk azokat. E modelleket és kombinációikat használják a klinikai immunológiában (Brusic et al., 2005) és az oltóanyag-kutatásban (Brusic – Petrovsky, 2005).
Konklúzió
A számítógépes modell a kísérleti kutatás fontos kiegészítő módszere. Különösképpen hasznosak azokon a területeken, melyek a rendszerek és folyamatok kombinatorikai jellegéből adódóan nagyszámú kísérletet igényelnek. Az ilyen területeken a számítógépes modellek az adatok mennyisége és az ismeret felhalmozódásával párhuzamosan alakulnak ki. Ebben a cikkben az immun epitópok tanulmányozásakor használt számítógépes modellek fejlődését írtuk le. A korai modellek, melyek kötési mintákra alapultak, durva szabályszerűségeket jeleztek a peptidadatbázisban. A következő generáció, a kötő mátrixok, a lineáris modelleket jelentik, melyek a peptid pozícióit számszerűsítve jellemzik. E modelleket kiszorították a kifinomult, nem lineáris modellek, melyek mesterséges neurális hálózatokat, rejtett Markov-modelleket vagy más eljárásokat használtak. A 3D elemzésen alapuló modellek kiegészítik az adatokra alapuló modelleket. Végül az egyedi modellek kombinálhatók az immunológiai „forró pontok”, a véletlen epitópok azonosításához, melyek az oltóanyag-kutatás legjobb célpontjai.
Kísérleti pozitívok Kísérleti negatívok Prediktált pozitívak Valódi pozitívak (VP) Hamis pozitívak (HP) Prediktált negatívak Hamis negatívak (HN) Valódi negatívak VN) Pontossági mutató Formula Párosul Érzékenység SE=TP/(TP+FN) SP Specificitás SP=TN/(TN+FP) SE Pozitív prediktív érték PPV= TP/(TP+FP) NPV Negatív prediktív érték NPV=TN/(TN+FN) PPV Keresztezési pont SE=SP - Pontosság Acc=(TP+TN)/(TP+TN+FP+FN) - Az ROC görbék integrálása (Swets, 1988) -
1. táblázat • Szakkifejezések meghatározása a prediktív modellek pontosságának megbecsülésében
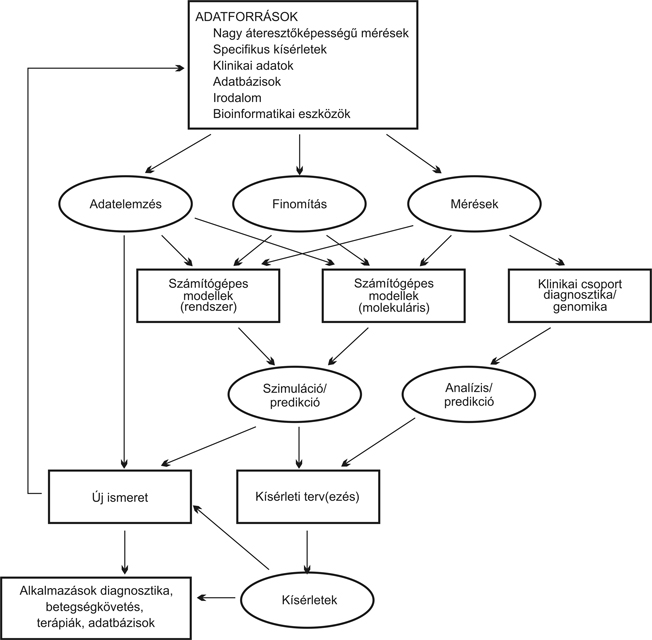
1. ábra • A számítógépes modellek kombinálása klinikai megfigyelésekkel és kísérletezéssel. Átdolgozva és bővítve (Brusic et al., 2005)-tól
Pozíció
1 2 3 4 5 6 7 8 9
Horgonyok F L
Y M
I
V
Kiegészít? horgonyok Y
Preferált aminosavak R N P R T N
I D I Q
L E E K
S K S
A T
2. táblázat • Kötődési minta az egér MHC molekulában H-2Kb. (Rammensee et al., 1999).
irodalom
Auffray, C. et al. (2003): From Functional Genomics to Systems Biology: Concepts and Practices. C. R Biol. 326, 10–11, 879–892.
Bajic, V. B. (2000): Comparing the Success of Different Prediction Software in Sequence Analysis: A Review. Brief Bioinform. 1, 3, 214–228.
Banvard, R. A. (2001): The Visible Human Project® Image Data Set from Inception to Completion and Beyond. Proceedings of CODATA 2002: Frontiers of Scientific and Technical Data. Track I-D-2: Medical and Health Data, Montréal, Canada, October, 2002.
Beer, M. A. – Tavazoie, S. (2004): Predicting Gene Expression from Sequence. Cell. 117, 2, 185–198.
Brusic, V. – Zeleznikow J. (1999): Computational Binding Assays of Antigenic Peptides. Lett Pept Sci. 6, 313–324.
Brusic, V. et al. (1998): Prediction of MHC Class II-Binding Peptides Using an Evolutionary Algorithm and Artificial Neural Network. Bioinformatics.14, 2, 121–130.
Brusic, V. et al. (2005). Information Technologies for Vaccine Research. Expert Rev Vaccines. 4, 407–417.
Brusic, V. – Petrovsky, N. (2005): Immunoinformatics and Its Relevance to Understanding Human Immune Disease. Expert Rev Clin Immunol. 1, 1, 145–157.
Cai, Y. D. et al. Support Vector Machines For Prediction of Protein Signal Sequences and Their Cleavage Sites. Peptides. 24, 1, 159–161.
Flower, D. R. (2003): Towards in Silico Prediction of Immunogenic Epitopes. Trends in Immunology. 24, 12, 667–674.
Fraser, C. M. – Rappuoli, R. (2005): Application of Microbial Genomic Science to Advanced Therapeutics. Annu Rev Med. 56, 459–474.
Hirschhorn, J. N. (2005): Genetic Approaches to Studying Common Diseases and Complex Traits. Pediatr Res. 57, 5 Pt 2:74R-77R.
Hunter, P. et al. (2005): Integration from Proteins to Organs: The IUPS Physiome Project. Mech Ageing Dev. 126, 1, 187–192.
Ishii, N, et al. (2004): Toward Large-Scale Modeling of the Microbial Cell for Computer Simulation. J. Biotechnol. 113, 1–3., 281–294.
Johnson, T. N. (2005): Modelling Approaches to Dose Estimation in Children. Br J Clin Pharmacol. 59, 6, 663–669.
Lador, F. et al. (2005): Simultaneous Determination of the Kinetics of Cardiac Output, Systemic O2 Delivery and O2 Lung Uptake at Exercise Onset in Men. Am J Physiol Regul Integr Comp Physiol.
Madden, D. R, et al. (1993): the Antigenic Identity of Peptide-MHC Complexes: A Comparison of the Conformations of Five Viral Peptides Presented by HLA-A2. Cell. 75, 4, 693–708.
Mamitsuka, H. (1998): Predicting Peptides That Bind to MHC Molecules Using Supervised Learning of Hidden Markov Models. Proteins. 33, 4, 460–474.
Matthews, L. – Woolhouse, M. (2005): New Approaches to Quantifying the Spread of Infection. Nat Rev Microbiol. 3, 7, 529–536.
Motta, S. – Brusic, V. (2004): Mathematical Modelling of the Immune System. In: Ciobanu, G. – Rozenberg, G. (eds.): Modelling in Molecular Biology. Natural Computing Series. Springer. 193–218.
Nielsen, H. et al. (1999): Machine Learning Approaches for the Prediction of Signal Peptides and Other Protein Sorting Signals. Protein Eng. Jan, 12, 1, 3–9.
Nowinski, W. L. et al. (2005): Informatics in Radiology (Inforad): Three-Dimensional Atlas of the Brain Anatomy and Vasculature. Radiographics. 25, 1, 263-271.
Petrovsky, N. – Brusic, V. (2004): Virtual Models of the HLA Class I Antigen Processing Pathway. Methods. 34, 4, 429–435.
Rammensee, H. G. et al. (1999): SYFPEITHI: Database for MHC Ligands and Peptide Motifs. Immunogenetics. 50, 3–4., 213–219.
Recanatini, M. et al. (2005): In Silico Modeling – Pharmacophores and Herg Channel Models. Novartis Found Symp. 266, 171–181.
Reifsnider, E. et al., (2005): Using Ecological Models in Research on Health Disparities. J Prof Nurs. 21, 4, 216–222.
Roberts, L. et al. (2001): A History of the Human Genome Project. Science. 291, 5507, 1195.
Muller, H. et al. 2004): A Review of Content-Based Image Retrieval Systems in Medical Applications-Clinical Benefits and Future Directions. Int J Med Inform. 73, 1, 1–23.
Schafroth, H. D. – Floudas, C. A. (2004): Predicting Peptide Binding to MHC Pockets Via Molecular Modeling, Implicit Solvation, and Global Optimization. Proteins. 54, 3, 534–556.
Scher, M. S. et al. (2005): Automated State Analyses: Proposed Applications to Neonatal Neurointensive Care. J. Clin Neurophysiol. 22, 4, 256–270.
Srinivasan, K. N. et al. (2004): Prediction of Class I T-Cell Epitopes: Evidence of Presence of Immunological Hot Spots inside Antigens. Bioinformatics. 20 Suppl I. I297–I302.
Suzuki, S. et al. (2005): Tele-Surgical Simulation System for Training in the Use of Da Vinci Surgery. Stud Health Technol Inform. 111, 543–548.
Swets, J. A. (1988): Measuring the Accuracy of Diagnostic Systems. Science. 240, 4857, 1285–1293.
Takahashi, K. et al. (2003): E-CELL2: Multi-Platform E-CELL Simulation System. Bioinformatics. 19, 13, 1727–1729.
Tompa, M. et al. (2005): Assessing Computational Tools for the Discovery of Transcription Factor Binding Sites. Nat Biotechnol. 23, 1,137–144.
Yu, K. et al. (2002): Methods for Prediction of Peptide Binding to MHC Molecules: A Comparative Study. Mol. Med. 8, 3, 137–148.
Yuan, Z. et al. (2004): Svmtm: Support Vector Machines to Predict Transmembrane Segments. J Comput. Chem. 25, 5, 632–636.
Zhao, Y. et al. (2003): Application of Support Vector Machines for T-Cell Epitopes Prediction. Bioinformatics. 19, 15, 1978–1984.
<-- Vissza a 2006/7 szám tartalomjegyzékére
<-- Vissza a Magyar Tudomány honlapra
[Információk] [Tartalom] [Akaprint Kft.]