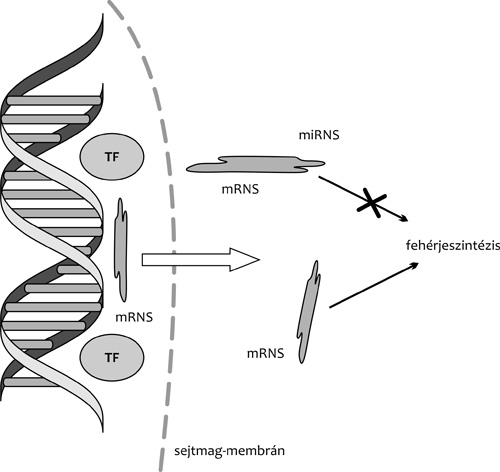
|
gének listáját ismerhetjük meg, hanem a gének
működésének egymáshoz való hálózati viszonyát is. Ennek révén sokkal
többet tudtunk meg a gének egymásra való közvetlen, vagy átkapcsoló
géneken keresztül, a géntermékeken át érvényesülő hatásáról, így
befolyásolhatóságáról is (Simkó et al., 2009).
A biológiai jelenségek komplexek, így genetikai
hátterük megismerése is komplex szemléletet követel. Egy bonyolult
jelátviteli folyamatban (pl. az inzulin hatása a sejtekre) tehát
minden komponens (csomópontok) ismerete mellett ezek egymáshoz való
viszonyának (élek) feltárása is szükséges. Betegségekben például
sokszor ebben a viszonyrendszerben következik be módosulás (3.
ábra).
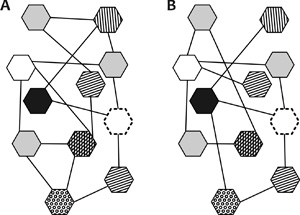
3. ábra • A hálózat elemei (például gének)
mellett a köztük lévő kapcsolatok is változhatnak A és B állapot
között.
A génhálózatok evolúciós jellegzetességeit két,
nagyon alapvető mechanizmus segítségével mutatjuk be.
Az egyik mindazon géneket tartalmazza, amelyek a
genom stabilitásának fenntartásáért felelősek. Ezek például a
DNS-hibák kijavításával foglalkozó gének (Kerzendorfer – O’Driscoll,
2009). Ez a mechanizmus teszi lehetővé az élő szervezetek generációkon
át megtartott alapvető sajátosságait.
A másik géncsoport az apoptózis-gének, amelyek a programozott
sejthalál bonyolult mechanizmusát szabályozzák (Elmore, 2007). Ezek a
gének, a sejt életének végén, annak zavarmentes elpusztulását és
eltávolítását irányítják. Ha ezek a gének a normálisnál fokozottabban
működnek, akkor a degeneratív folyamatok dominálnak (például
Alzheimer-kór), ha csökkent a működés, akkor a daganatos betegségek
valószínűsége nő.
Az elmúlt pár évben tudtuk meg, hogy mindkét
rendszer sok gént tartalmaz, melyek hálózatban,
sokszorosan szabályozott, bonyolult együttműködésben működnek.
Mintegy száz apoptózis-gén és kb. nyolcvan
genomstabilitás-gén „kapcsolatrendszerének” vizsgálata során kiderült
(Castro et al., 2008), hogy a genomstabilitás és az apoptózis
szegmenseken belül külön-külön is több száz génkölcsönhatás érvényesül
(4. ábra), de a két rendszer egymással is jelentős számú
közvetlen génkölcsönhatási kapcsolatban áll. Ráadásul a törzsfejlődés
egyes állomásain található élő rendszerek genomstabilitási és
sejthalál génhálózatai jelentős evolúción mentek át.
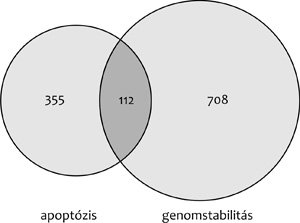
4. ábra • A körökben lévő számok az
apoptózis- és a genomstabilitás-gének csoportján belüli illetve a
közös gén–gén kapcsolatokat jelzi, a körök nagysága ezek arányára
utal.
Ha az evolúciós változásokat egymáshoz hasonlítjuk,
e két rendszerben kitűnik azok koevolúciós tendenciája.
Az evolúciós vizsgálat olyan bioinformatikai
eszközrendszerrel történik, ahol az eljárás képes az egymásnak
megfelelő szerkezetű és funkciójú géneket („ortológokat”)
kiválasztani, és a hálózatban együtt kezelni.
Ha az így „normalizált” genomstabilitás/apoptózis
hálózatokat a maláriát okozó Plasmodium parazita, a Caenorhabditis
elegans nevű fonálféreg és az ember viszonylatában hasonlítjuk össze,
egyértelműen kiderül, hogy a genomstabilitás rendszer az ősibb, az
evolúció során viszonylag kevés változást mutat. Ugyanakkor az
apoptózis hálózatok evolúciója nagyon jelentős. A mitokondriális
eredetű apoptózis gének a legősibbek, hiszen ezek már az egysejtűnél
is megtalálhatók, az endogén apoptózis mechanizmusokban szereplő egyes
enzimek (kaszpáz) a fonalférgeknél kimutathatóak, míg a külső
hatásokra beinduló sejthalál-kaszkádban döntő faktorok (pl. tumor
nekrózis faktor) e három egymástól evolúciósan messze álló élő
rendszerben csak az emberi apoptózis-gének között lelhető fel.
Ebben a rendszerben lehetőség nyílik az egyes génhálózatok
plaszticitásának vizsgálatára is. A plaszticitás itt azt jelenti, hogy
egyes nagy, drámai hatások (például egy sok gént érintő deléció,
duplikáció vagy akár átrendeződés) nyomán miképp áll helyre a működés,
például a genomstabilitás és apoptózis rendszer. Ez a szituáció
transzgenikus állatmodelleken gének, géncsoportok „kiütésével”
(knock-out) is szimulálható. Ezek a vizsgálatok is a
genomstabilitás-gének nagyfokú, a sejthalál-gének kisebb mértékű
konzervativizmusát igazolták.
Ezzel a hálózatos ábrázolással sikerült azt is
kimutatni, hogy emberi tumorokban a genomstabilitás-gének változatai
inkább öröklődő, míg az apoptózis-rendszer komponenseinek sokféleségét
inkább az egyedi élet során (például környezeti hatásokra) kialakuló
mutációk alakítják ki.
A génhálózatok kialakulásának, kapcsolati
szövedékének, plaszticitásának megismerése többek között arra ad majd
lehetőséget, hogy új gyógyszerek tervezésénél a megfelelő célpontokat
válasszák ki a betegségek eredményesebb kezelése érdekében. Ez a
genetikusok, orvosok, bioinformatikusok közötti szorosabb
együttműködést igényli és teszi lehetővé a közeli jövőben.
Kulcsszavak: gén, génhálózat, nem átíródó RNS-szekvenciák,
mikro-RNS, transzkripciós faktor, genomstabilitás, programozott
sejthalál
IRODALOM
Caldas, Carlos – Brenton, James D. (2005):
Sizing up miRNAs as cancer genes. Nature Medicine. 11, 712–714.
Castro, Mauro A. A. – Dalmolin, R. J. –
Moreira, J. C. – Mombach, J. C. – de Almeida, R. M. (2008):
Evolutionary Origins of Human Apoptosis and Genome-stability Gene
Networks. Nucleic Acids Research. 36, 19, 6269–283.
WEBCÍM >
Elmore, Susan (2007): Apoptosis: A Review
of Programmed Cell Death. Toxicologic Pathology. 35, 4, 495–516.
WEBCÍM >
Feuer, Michael J. – Towne, L. – Shavelson,
R. J. (2002): Scientific Culture and Educational Research. The
Educational Researcher. 31, 8, 4–14. (újraközlés):
WEBCÍM >
Hua, Zhong – Lv, Q. – Ye, W. – Wong, C. K.
– Cai, G. (2006): MiRNA-Directed Regulation of VEGF and Other
Angiogenic Factors under Hypoxia. PLoS ONE. 1:e116.
WEBCÍM >
Kerzendorfer, Claudia – O’Driscoll, Mark
(2009): Human DNA Damage Response and Repair Deficiency Syndromes:
Linking Genomic Instability and Cell Cycle Checkpoint Proficiency. DNA
Repair (Amst). 8, 9, 1139–1152.
Lander, Eric S. et al. (252 coauthors)
International Human Genome Sequencing Consortium (2001): Initial
sequencing and analysis of the human genome. Nature, 409, 6822,
860–921.
WEBCÍM
>
Sevignani, Cinzia – Calin, G. A. –
Siracusa, L. D. – Croce, C. M. (2006): Mammalian microRNAs: A Small
World for Fine-Tuning Gene Expression. Mammalian Genome. 17, 189–202.
WEBCÍM
>
Simkó Gábor I. – Gyurkó D. – Veres D. V. –
Nánási T. – Csermely P. (2009):
Network Strategies to Understand the Aging Process and Help
Age-related Drug Design. Genome Medicine. 1, 9, 90.
Valencia-Sanchez, Marco Antonio – Liu, J.
– Hannon, G. J. – Parker, R. (2006): Control of Translation and mRNA
Degradation by miRNAs and siRNAs. Genes Dev. 20, 515–24.
WEBCÍM >
Venter, J. Craig et al (228 coauthors)
(2001): The Sequence of the Human Genome. Science. 291, 5507,
1304–1351.
WEBCÍM
>
|