|
Úton a pontosabb faj- és géntörténetek felé
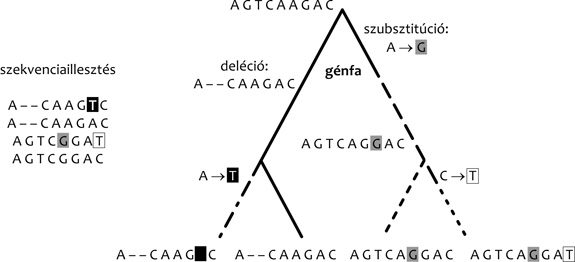
Mint azt fentebb is említettük, a géncsaládok szekvenciája alapján
rekonstruált gének gyakran elmosódottak. Ha a fajfát ismerjük, vagy
ismertnek tételezzük fel, nagy mértékben csökkenteni tudjuk ezt az
elmosódottságot. Ekkor ugyanis a lehetséges génfák közül nem csak
aszerint tudunk választani, hogy melyeket feltételezve van
szükségünk a legkevesebb szubsztitúcióra (vö. 1. ábra), hanem
az alapján is, melyik génfa magyarázható kisebb számú duplikáció,
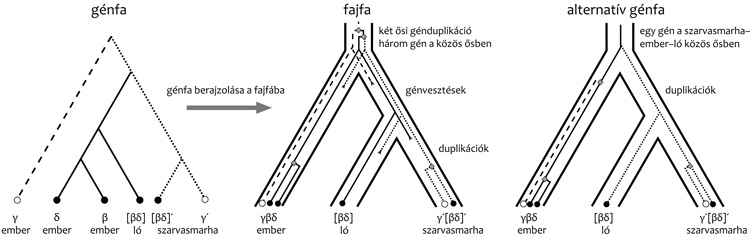
horizontális géntranszfer és génvesztés segítségével. A 2. ábra
példája esetén is ez a helyzet, kiderül ugyanis, hogy a fajfa
ismeretében egy alternatív génfa lesz a legvalószínűbb, amelynek a
fajfába való berajzolásához kevesebb duplikáció- és
génvesztés-eseményre van szükség.
De honnan tudjuk, hogyan néz ki a fajfa? A
β-globinokon kívül géncsaládok millióit ismerjük. Ezek közül azokban
a géncsaládokban, amelyekben duplikáció-, génvesztés- vagy
transzferesemények mentek végbe, azt várjuk, hogy a génfa különböző
lesz a fajfától. Valójában a géncsaládok szinte mindegyike ebbe a
kategóriába sorolható, olyan eszenciális géncsaládok ritka
kivételével, amelyek hiánya drasztikus következményekkel jár az
élőlények számára – ilyen például az információkezeléssel
(transzkripció, transzláció) foglalkozó gének egy része. Amennyiben
egy ilyen géncsalád tagjai pontosan egy példányban szerepelnek
minden élőlény genomjában, és feltételezzük, hogy nem történt
egyetlen génduplikáció, horizontális géntranszfer vagy
génvesztés-esemény sem, akkor a géncsalád története, pontosabban az
azt leíró génfa megegyezik az őket hordozó fajok történelmével,
vagyis az azt leíró fajfával. 1977-ben Carl Woese és társai (Fox et
al., 1977) is egy ilyen „egypéldányos” géncsalád, a riboszómák
alkotórészét adó kis RNS-alegység, az ún. 16S rRNS gének
szekvenciáit vizsgálva rekonstruálták az élővilág fajfáját, és
fedezték fel, hogy az addig prokariótaként ismert csoport valójában
két doménre oszlik: a baktériumokra és archeákra.
Célravezetőnek tűnhet tehát, hogy a kis számú
esszenciális géncsaládot, a gének „1%”-át felhasználva kövessük
vissza az élet történetét. Azonban ha így járunk el, a géncsaládok
maradék 99%-a által hordozott információt nem hasznosítjuk. Ez azt
eredményezi, hogy az egyedi géncsaládok történetének elmosódottsága
miatt sok esetben nem rajzolható fel egyértelműen a fajfa sem.
Erre a problémára keresték a megoldást Bastien
Boussau és munkatársai, akik 2013-ban harminchat emlősfajban 6966
géncsalád történetét rekonstruálták a fajfával közösen. A lehetséges
fajfák terét egy számítógépes algoritmus segítségével járták be: egy
adott fajfára elkészítették az összes géncsaládhoz tartozó, az adott
fajfához és a szekvenciákhoz együttesen legjobban illő, mindkét
folyamat szerint legvalószínűbb génfát. Az így kapott „közös”
valószínűségek szorzata adta a fajfa valószínűségét. Ezt követően
minden szomszédos fajfára is elvégezték ugyanezt a számítást, és ha
ezek között találtak olyan szomszédot, melynek valószínűsége nagyobb
volt, mint a kezdeti fajfa, akkor az eljárást ettől a fajfától
elindulva megismételték. Az algoritmus akkor ért véget, amikor olyan
fajfát találtak, amelynek nem volt szomszédja, amely nála
valószínűbb lett volna. Ez az eljárás egyszerre használja a fajfa
adta keretrendszert arra, hogy pontosabb génfákat kapjunk, miközben
a fajfát is nagy pontossággal, több ezer géncsalád alapján képes
meghatározni — ez az ún. közös rekonstrukció. Az eredmények
biztatóak: a kapott fajfa nem mond ellent semmilyen ismert
rendszertani ténynek, és a géntörténetek is valószerűek, ugyanis nem
mutatnak a mainál jóval nagyobb ősi genomokat (nem megfelelő
rekonstrukciós módszerek gyakran vezetnek „felduzzasztott” ősi
genomokhoz).
Géntranszfer: zaj helyett információ
Az emlősök történetének kutatásában elegendő csupán a duplikációkat
és a génvesztéseket figyelembe vennünk, más élőlényeknél viszont nem
hanyagolható el a gének történetét bonyolító harmadik folyamat sem:
a horizontális géntranszfer. A transzfereseményekre sokan a fajfa
vonalait elmosó „zajként” tekintenek, hiszen ilyenkor a génfák
vonalai átlépik a fajok szabta határokat, és a fajfa csöveinek
összekötésével egy bonyolult hálózatot hoznak létre. Ha azonban
mégis arra vállalkozunk, hogy megvizsgáljuk a transzferen
keresztülment géncsaládok történetét is, értékes információkhoz
juthatunk.
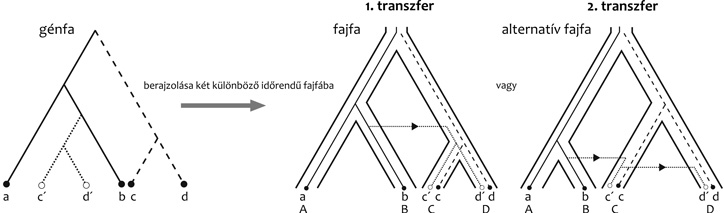
Először is figyelembe kell vennünk, hogy minden
transzfer két, térben és időben egymás mellett élő faj között
történhet meg – fordított szemszögből nézve, ha ismerünk egy
transzfert is tartalmazó géntörténetet, megtudhatjuk, a fajfa mely
ágai fedtek át egymással időben (3. ábra). Több transzfert
felhasználva akár a fajfa összes elágazásának idejét is
meghatározhatjuk (Szöllősi et al., 2012).
Ugyanilyen jelentőséggel bírhat a tény, hogy az
ősi horizontális transzferek során mára már kihalt fajok génjei is
bekerülhettek a mostani élőlények őseinek genomjába. Ha ezek a gének
beépültek az új genomba, és ott egészen napjainkig fennmaradtak,
akkor az élővilág számunkra eddig ismeretlen részeinek lenyomatát
adhatják (Szöllősi et al., 2013).
A múlt életre kel a laboratóriumban
A filogenetikai rekonstrukció eredménye nemcsak egy génfa, hanem,
mint az az 1. ábrán is látható, a szekvenciaváltozásokat a
génfa mentén visszafelé követve az ősi szekvenciákat is megkapjuk.
Ugyanazon szekvenciaillesztés esetén különböző alternatív génfák
különböző ősi szekvenciákhoz vezetnek. Ezek az ősi szekvenciák az
evolúciós múltról tett alternatív predikciók, amelyeket a
laboratóriumban egymással összehasonlíthatunk. Rendelkezésünkre
állnak ugyanis molekuláris biológiai módszerek, melyek segítségével
az ősi szekvenciákat mesterségesen szintetizálva, majd baktériumokba
bejuttatva az általuk kódolt ősi fehérjéket is legyárthatjuk.
Mathieu Groussin és társai (Groussin et al.,
2015) ilyen módon „feltámasztott,” több száz millió éves
LeuB-enzimek biokémiai tulajdonságait hasonlították össze. Arra az
eredményre jutottak, hogy mind a fajfa figyelembe vételével, mind
pedig az anélkül rekonstruált génfáknak megfelelő enzim nagy
vonalakban hasonló enzimeket eredményezett: mindkettő a mai
LeuB-enzimeknél magasabb hőmérsékleten működött optimálisan,
megerősítve azt, hogy az ősi baktérium, amelyből származtak, magas
hőmérsékleteken élő, ún. termofil organizmus volt. A részletesebb
enzimatikus aktivitásra irányuló vizsgálatok azonban egyértelműen
kimutatták, hogy csak a fajfa felhasználásával rekonstruált enzim
mutatott a jelenbeli enzimekkel összemérhető, valószerű biokémiai
paramétereket.
A LeuB-enzimekkel végzett kísérletek közvetlen
bizonyítékát adják annak, hogy a fajfa és génfák közös kezelésével
pontosabb rekonstrukciókat kapunk. Megmutatják továbbá, hogy nemcsak
a fajok és gének leszármazásának történetéről lehet élesebb
fogalmunk, hanem az ősi enzimekről és rajtuk keresztül az ősi
élőlények tulajdonságairól is részletes információkhoz juthatunk.
Kulcsszavak: DNS-szekvenálás, génduplikáció, horizontális
géntranszfer, szekvenciaillesztés, génfák, fajfák
IRODALOM
Boussau, Bastien – Szöllősi Gergely J. –
Duret, Laurent et al. (2013): Genome-scale Coestimation of Species
and Gene Trees. Genome Research. 23, 2, 323–330. DOI: 10.1101/
gr.141978.112 •
WEBCÍM
Boussau, Bastien – Daubin, Vincent
(2010): Genomes as Documents of Evolutionary History. Trends in
Ecology and Evolution. 25, 4, 224–232. DOI: 10.1016/j.tree.
2009.09.007
Felsenstein, Joseph (2004): Inferring
Phylogenies. Sinauer Associates, Sunderland, USA
Fox, George E. – Magrum, Linda J. –
Balch, William E. et al. (1977): Classification of Methanogenic
Bacteria by 16S Ribosomal RNA Characterization. Proceedings of the
National Academy of Sciences of the USA. 74, 10, 4537–4541. DOI:
10.1073/ pnas.74.10. 4537 •
WEBCÍM
Groussin, Mathieu – Hobbs, Joanne K. –
Szöllősi Gergely J. et al. (2015): Toward More Accurate Ancestral
Protein Genotype–Phenotype Reconstructions with the Use of Species
Tree-aware Gene Trees. Molecular Biology and Evolution. 32, 1,
13–22. DOI: 10.1093/ molbev/msu305
Maddison, Wayne P. (1997): Gene Trees in
Species Trees. Systematic Biology. 46, 3, 523–536. DOI:
10.1093/sysbio/46.3.523 •
WEBCÍM
Szöllősi Gergely J. – Tannier, Eric –
Lartillot, Nicolas – Daubin, Vincent (2013): Lateral Gene Transfer
from the Dead. Systematic Biology. 62, 3, 386–397. DOI:
10.1093/sysbio/syt003 •
WEBCÍM
Szöllősi Gergely J. – Boussau, Bastien –
Abby, Sophie S. et al. (2012): Phylogenetic Modeling of Lateral Gene
Transfer Reconstructs the Pattern and Relative Timing of
Speciations. Proceedings of the National Academy of Sciences of the
USA. 109, 43, 17513–17518. DOI: 10.1073/pnas.1202997109 •
WEBCÍM
Zuckerkandl, Emile – Pauling, Linus
(1965): Evolutionary Divergence and Convergence in Proteins.
Evolving Genes and Proteins. 97, 97–165. •
WEBCÍM
Zuckerkandl, Emile – Pauling, Linus
(1965): Molecules as Documents of Evolutionary History. Journal of
Theoretical Biology. 8, 2, 357–366. DOI:
110.1016/0022-5193(65)90083-4 •
WEBCÍM
|