|
aktivációt eredményező agonisták ([Sar1, Ile4,
Ile8]-AngII) használatával is. Kimutattuk, hogy az AngII hatásával
ellentétben [Sar1, Ile4, Ile8]-AngII agonista hatására a receptor nem
jelenik meg a nem-raft mikrodoménben a receptor internalizációját
megelőzően HEK293-sejtekben (Balla et al, 2012). E vizsgálatok
alátámasztják, hogy a receptorok bizonyos mutációi, illetve egyes
agonisták képesek a receptor jelátviteli folyamatainak szelektív
aktiválására.
Betegséget okozó mutáció azonosítása
a V2-receptorban
A Semmelweis Egyetem II. Belgyógyászati Klinika munkatársaival (Dr.
Patócs Attila, Dr. Tóth Miklós, Prof. Dr. Rácz Károly) együttműködve
azonosítottunk egy nefrogén diabétesz inszipidusz betegben előforduló
missense mutációt a V2-R génjében. Kísérleteinkben arra kerestük a
választ, hogy a betegben azonosított mutáció a receptor mely
funkcióját károsítja; a továbbiakban olyan V2-R ligandokat próbálunk
azonosítani, amelyek a receptor jelátviteli folyamatait szelektíven
aktiválják, és amelyek emiatt pozitívan befolyásolják a mutáns
receptor működését. Első lépésben vizsgáltuk a mutáns receptor
működését, mert nem állt rendelkezésre irodalmi adat arról, hogy e
rendkívül ritka mutáció milyen mechanizmussal okoz funkcióvesztést. A
méréseink során élősejtes kísérletekben vizsgáltuk a mutációt
tartalmazó receptor tulajdonságait, és összevetettük őket a normális
(eredeti) V2-receptor tulajdonságaival.
Kísérleteink elvégzése céljából olyan
tesztrendszereket fejlesztettünk ki, melyek alkalmasak lehetnek
nagyszámú vizsgálat (HTS) elvégzésére is. Erre alkalmas a nagy
érzékenységű, 96-lyukú lemezeken, élő sejtekben is elvégezhető
BRET-módszer. A V2-R olyan GFKR, mely Gs típusú heterotrimer
G-fehérjét képes aktiválni. A különböző Gs-fehérje kapcsolt receptorok
a fiziológiás agonista kötését követően elsősorban a Gs heterotrimer
G-fehérje által közvetített jelátviteli útvonalakat aktiválnak, és
adenilát-cikláz aktiválásán keresztül cAMP-függő jelátviteli
folyamatok indulnak el. Kimutatták, hogy ezen típusú receptorok más
jelátviteli útvonalakat is aktiválhatnak, melyek közül fontosnak tűnik
a β-arresztin-kötés és a MAP-kináz (mitogén-aktivált protein-kináz)
kaszkád elindítása emlős sejtekben. Megvizsgáltuk a mutáns receptor
tulajdonságait, és összevetettük az eredeti receptoréval. Konfokális
mikroszkópos eredmények alapján kimutattuk, hogy a mutáns receptor
megtalálható a sejtmembránban, és a receptorszám az eredeti
receptoréval összevethető nagyságrendű. További kísérleteinkben a
sejten belüli cAMP-szint mérését végeztük el, mivel a V2-R-agonista
kötése ezt a másodlagos hírvivő képzését indítja el. A HTS-vizsgálatok
kivitelezhetőségét szem előtt tartva, alkalmazhatunk olyan rezonancia
energiatranszferen alapú bioszenzorokat, amelyek segítségével
élősejtes kísérletekben nyomon követhetjük a receptor aktiválódását. A
cAMP a szabályozó hatását fehérjékhez kapcsolódva fejti ki, ilyen
fehérje az EPAC (Exchange Protein directly Activated by cAMP), amely
kis G-fehérjék működését befolyásolja. Az EPAC-fehérje cAMP-kötését
követően konformációváltozáson megy keresztül, megteremtve a cAMP
szenzorként való alkalmazásának lehetőségét. Az irodalomban már
leírtak egy EPAC-alapú cAMP-szondát, amelynek működése fluoreszcencia
rezonancia energiatranszferen (FRET) alapult (Ponsioen et al, 2004). A
FRET-módszer hátránya a magas jel/zaj arány, valamint
korlátozott alkalmazhatósága nagy elemszámú méréseknél, így
szükségessé vált egy érzékenyebb módszer kidolgozása. Molekuláris
biológiai módszerekkel biolumineszcens luciferáz fehérjét építettünk a
bioszenzorba, amely már BRET-alapú, nagy érzékenységű, valós idejű
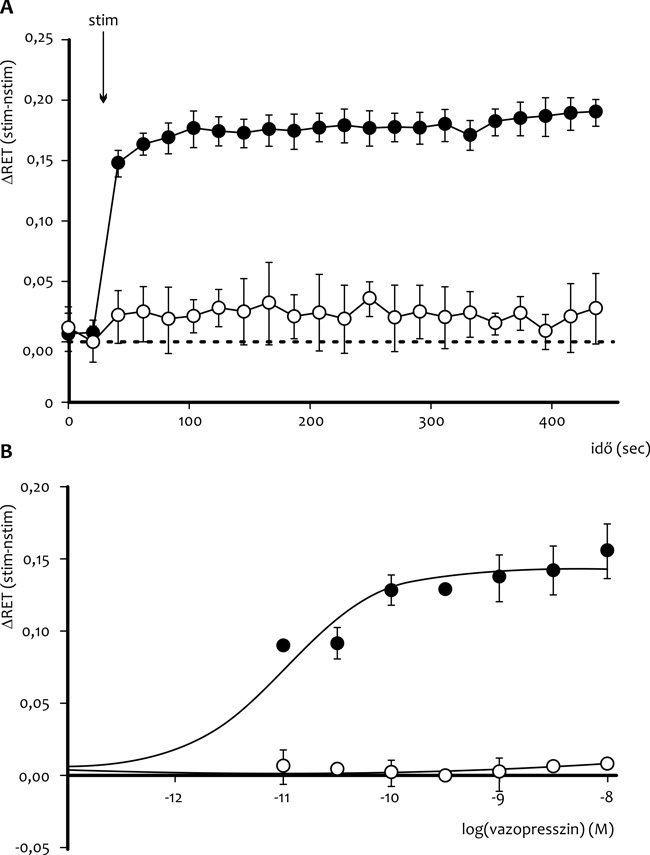
mérések kivitelezését tett lehetővé (1. ábra,
A). Ezen új bioszenzor felhasználásával megvizsgáltuk a mutációt
tartalmazó V2-R cAMP jelátvitelét HEK293-sejtekben. Vazopresszin
hatására a vad típusú receptordózis-függő cAMP válaszát detektáltuk,
de a mutáns receptor fiziológiás hormonszint-tartományban nem hozott
létre cAMP-szint emelkedést (1 ábra, B). Mivel a vazopresszin
V1-receptoron keresztüli érszűkítő (és vérnyomásemelő) hatást hozhat
létre, ezért a terápiában dezmopresszint alkalmaznak, mely egy
V2R-szelektív vazopresszin analóg. Kísérleteinkben a dezmopresszin még
nagy dózisban alkalmazva sem volt képes aktiválni a mutáns receptort.
A mutáció hatása a receptor jelátvitelére
További kísérleteinkben megvizsgáltuk, hogy a különböző Gs-kapcsolt
receptorok létrehozzák-e a sejtproliferáció, sejtnövekedés és számos
egyéb transzkripciós szintű szabályozásában központi szerepet játszó
Ras-aktiválódást, illetve az ennek következtében létrejövő Ras- kis
G-fehérje-aktiválódását. Gs-kapcsolt β2 adrenerg receptort (β2-R), az
MC melanokortin receptort (MC4-R), az 5HT-7A szerotonin receptort
(5HT7A-R) és a V2 receptorokat expresszáltunk HEK293-sejtekben, és
megmértük a cAMP-jelet, a MAPK-foszforilációt és a Ras-aktiválódást
(Balla et al., 2011). A sejtek endogén receptorainak feltérképezésére
receptorral nem transzfektált sejteket is használtunk. Forskolin
használatával, ami a cAMP képzéséért felelős enzimet közvetlenül
aktiválja, a receptorok agonista kötése nélkül válthattunk ki
cAMP-jelet, és az így kapott adatokat összevetettük a receptorokkal
kapott adatokkal. Eredményeink szerint a V2-R a Ras által kiváltott
jelpályán keresztül, míg a forskolin ingerlés, illetve a β2-R-, az
MC4-R- és az 5HT7A-R-hormon kötése Ras-független útvonalon vált ki
MAPK aktiválódást. Az általunk azonosított, betegséget okozó mutációt
tartalmazó V2 receptort alkalmazva viszont nem kaptunk Ras-jelpálya
aktiválódást, ami alátámasztja, hogy a receptor mutációja
következtében a jelátviteli folyamat károsodott. Előzetes kísérleteink
arra utalnak, hogy nagy dózisú vazopresszin képes aktiválni az
általunk vizsgált mutáns receptort, ezért további kísérleteink célja
olyan receptor agonisták azonosítása, melyek képesek e mutáns receptor
aktiválására.
Összefoglalás
Kísérleteinkben kimutattuk, hogy az általunk használt megközelítési
módok alkalmasak GFKR-ok aktivációjának és szelektív jelátvitelének
tanulmányozására, valamint segítséget nyújthatnak a betegséget okozó
receptormutációk jobb megértéséhez. Vizsgálataink azonosítottak egy
betegséget okozó V2-R-mutációt. További kísérleteinkben olyan peptid-
és nem-peptid vazopresszin receptor ligandokat próbálunk azonosítani,
amelyek az általunk meghatározott, károsodott receptor mutáns
működését kedvezően befolyásolják. Az általunk beállított
BRET-technikán alapuló tesztrendszerek alkalmasak nagyszámú minta
analízisére, így van esély arra, hogy vegyületkönyvtárak
felhasználásával olyan molekulákat találjunk, melyek a mutáns V2-R
működését kedvezően befolyásolják.
A nefrogén diabétesz inszipidusz betegséget számos más mutáció
létrehozhatja, így az alkalmas terápiás megoldásnak a mutációtól
függően egyénre szabottnak kell lennie. Ebben segíthet a különböző,
betegséget okozó receptor mutációk működésének megértése és olyan
gyógyszerjelöltek azonosítása, melyek az adott receptormutációt
hordozó betegeken segíthetnek.
Kulcsszavak: G-fehérje kapcsolt receptorok, vazopresszin receptor,
biolumineszcencia energiatranszfer (BRET), szelektív jelátvitel,
személyre szabott terápia
IRODALOM
Balla A. – Erdélyi L. S. – Soltész-Katona
E. – Balla T. – Várnai P. – Hunyady L. (2011): Demonstration of
Angiotensin II-Induced Ras Activation in the Trans-Golgi Network and
Endoplasmic Reticulum Using Bioluminescence Resonance Energy
Transfer-Based Biosensors. The Journal of Biological Chemistry. 286,
5319–5327.
Balla A. – Tóth D. – Soltész-Katona E. –
Szakadáti G. – Erdélyi L. S. – Várnai P. – Hunyady L. (2012): Mapping
of the Localization of Type I Angiotensin Receptor in Membrane
Microdomains Using Bioluminescence Resonance Energy Transfer-Based
Sensors. The Journal of Biological Chemistry. 287, 9090–9099.
Ferguson, S. S. (2001): Evolving Concepts
in G Protein-Coupled Receptor Endocytosis: The Role in Receptor
Desensitization and Signaling. Pharmacological Reviews. 53, 1–24.
Hunyady L. – Catt, K. J. (2006):
Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological
and Pathogenic Actions of Angiotensin II. Molecular Endocrinology. 20,
5, 953–970.
Jean-Alphonse, F. – Perkovska, S. –
Frantz, M. C. – Durroux, T. – Mejean, C. – Morin, D. – Loison, S. –
Bonnet, D. – Hibert, M. – Mouillac, B. – Mendre, C. (2009): Biased
Agonist Pharmacochaperones of the AVP V2 Receptor May Treat Congenital
Nephrogenic Diabetes Insipidus. Journal of the American Society of
Nephrology. 20, 2190–2203.
Kenakin, T. – Miller, L. J. (2010): Seven
Transmembrane Receptors As Shapeshifting Proteins: the Impact of
Allosteric Modulation and Functional Selectivity on New Drug
Discovery. Pharmacological Reviews 62, 265–304.
Kenakin, T. (2011): Functional Selectivity
and Biased Receptor Signaling. The Journal of Pharmacology and
Experimental Therapeutics 336, 296–302.
Ma, P. – Zemmel, R. (2002): Value of
Novelty? Nature Reviews 1, 571–572.
Ponsioen, B. – Zhao, J. – Riedl, J. –
Zwartkruis, F. – Van Der Krogt, G. – Zaccolo, M. – Moolenaar, W. H. –
Bos, J. L. – Jalink, K. (2004): Detecting cAMP-Induced Epac Activation
by Fluorescence Resonance Energy Transfer: Epac as a Novel cAMP
Indicator. EMBO Reports. 5, 12,1176–1180.
Rajagopal, S. – Rajagopal, K. – Lefkowitz,
R. J. (2010): Teaching Old Receptors New Tricks: Biasing
Seven-Transmembrane Receptors. Nature Reviews. 9, 373–386.
Spanakis, E. – Milord, E. – Gragnoli, C.
(2008): AVPR2 Variants and Mutations in Nephrogenic Diabetes
Insipidus: Review and Missense Mutation Significance. Journal of
Cellular Physiology. 217, 605–617.
Szidonya L. – Süpeki K. – Karip, E. –
Turu, G. – Várnai P. – Clark, A. J. – Hunyady L. (2007): AT1 Receptor
Blocker-Insensitive Mutant AT1A Angiotensin Receptors Reveal the
Presence of G Protein-Independent Signaling in C9 Cells. Biochemical
Pharmacology. 73, 10,1582–1592.
Wei, H. – Ahn, S. – Shenoy, S. K. –
Karnik, S. S. – Hunyady L. – Luttrell, L. M. – Lefkowitz, R. J.
(2003): Independent Beta-Arrestin 2 and G Protein-Mediated Pathways
for Angiotensin II Activation of Extracellular Signal-Regulated
Kinases 1 and 2. Proceedings of the National Academy of Sciences of
the United States of America 100, 10782–87.
|